Medical expert of the article

New publications
Pulmonary hypertension
Last reviewed: 12.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
Pulmonary hypertension (pulmonary arterial hypertension) is an increase in pressure in the pulmonary artery system, which may be due to an increase in resistance in the pulmonary vascular bed or a significant increase in the volume of pulmonary blood flow. This pathology is secondary in most cases; when the cause is unknown, it is called primary. In primary pulmonary hypertension, the pulmonary vessels narrow, hypertrophy, and fibrosis.
Pulmonary hypertension results in right ventricular overload and failure. Symptoms of pulmonary hypertension include fatigue, shortness of breath on exertion, and sometimes chest discomfort and fainting. Diagnosis is made by measuring pulmonary artery pressure. Treatment of pulmonary hypertension includes vasodilators and, in some severe cases, lung transplantation. The prognosis is generally poor unless a treatable cause is identified.
Normally, the pressure in the pulmonary artery is:
- systolic - 23-26 mm Hg
- diastolic - 7-9 mm Hg
- average -12-15 mm Hg
According to WHO recommendations, the upper limit of the norm for systolic pressure in the pulmonary artery is 30 mm Hg, diastolic - 15 mm Hg.
Causes pulmonary hypertension
Pulmonary hypertension occurs when the mean pulmonary arterial pressure is > 25 mmHg at rest or > 35 mmHg during exercise. Many conditions and medications cause pulmonary hypertension. Primary pulmonary hypertension is pulmonary hypertension in the absence of such causes. However, the outcome may be similar. Primary pulmonary hypertension is rare, with an incidence of 1 to 2 people per million.
Primary pulmonary hypertension affects women twice as often as men. The average age at diagnosis is 35 years. The disease may be familial or sporadic; sporadic cases are approximately 10 times more common. Most familial cases have mutations in the bone morphogenetic protein receptor type 2 (BMPR2) gene, a member of the transforming growth factor (TGF)-beta receptor family. Approximately 20% of sporadic cases also have BMPR2 mutations. Many people with primary pulmonary hypertension have elevated levels of angioprotein 1; angioprotein 1 appears to down-regulate BMPR1A, a BMPR2-related protein, and may stimulate serotonin production and endothelial smooth muscle cell proliferation. Other possible associated factors include disorders of serotonin transport and human herpesvirus 8 infection.
Primary pulmonary hypertension is characterized by variable vasoconstriction, smooth muscle hypertrophy, and vessel wall remodeling. Vasoconstriction is thought to be due to increased thromboxane and endothelin 1 activity (vasoconstrictors) on the one hand, and decreased prostacyclin and nitric oxide activity (vasodilators) on the other. Increased pulmonary vascular pressure, which occurs due to vascular obstruction, worsens endothelial damage. Damage activates coagulation on the intimal surface, which can worsen hypertension. This may also be facilitated by thrombotic coagulopathy due to increased levels of plasminogen activator inhibitor type 1 and fibrinopeptide A and decreased activity of tissue plasminogen activator. Focal coagulation on the endothelial surface should not be confused with chronic thromboembolic pulmonary arterial hypertension, which is caused by organized pulmonary thromboemboli.
Ultimately, in most patients, primary pulmonary hypertension leads to right ventricular hypertrophy with dilation and right ventricular failure.
The causes of pulmonary hypertension are presented in the classification.
 [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
[ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
Etiological classification of pulmonary hypertension
Left ventricular failure
- Ischemic heart disease.
- Arterial hypertension.
- Aortic valve defects, coarctation of the aorta.
- Mitral regurgitation.
- Cardiomyopathy.
- Myocarditis.
Increased pressure in the left atrium
- Mitral stenosis.
- Tumor or thrombosis of the left atrium.
- Triatrial heart, supravalvular mitral annulus.
Pulmonary vein obstruction
- Mediastinal fibrosis.
- Pulmonary venous thrombosis.
Parenchymatous lung diseases
- Chronic obstructive pulmonary diseases.
- Interstitial lung diseases (disseminated processes in the lungs).
- Acute severe lung injury:
- adult respiratory distress syndrome;
- severe diffuse pneumonitis.
Diseases of the pulmonary artery system
- Primary pulmonary hypertension.
- Recurrent or massive pulmonary embolism.
- In situ thrombosis of the pulmonary artery.
- Systemic vasculitis.
- Distal pulmonary artery stenosis.
- Increased pulmonary blood flow:
- congenital heart defect with left-to-right shunting (ventricular septal defect, atrial septal defect);
- patent ductus arteriosus.
- Drug- and food-induced pulmonary hypertension.
Pulmonary hypertension in newborns
- Preserved fetal circulation.
- Hyaline membrane disease.
- Diaphragmatic hernia.
- Meconium aspiration.
Hypoxia and/or hypercapnia
- Living in high mountain areas.
- Upper airway obstruction:
- enlarged tonsils;
- obstructive sleep apnea syndrome.
- Obese hypoventilation syndrome (Pickwickian syndrome).
- Primary alveolar hypoventilation.
Many authors consider it appropriate to classify pulmonary hypertension depending on the time of its development and to distinguish between acute and chronic forms.
Causes of Acute Pulmonary Hypertension
- PE or thrombosis in situ in the pulmonary artery system.
- Acute left ventricular failure of any origin.
- Asthmatic status.
- Respiratory distress syndrome.
Causes of Chronic Pulmonary Hypertension
- Increased pulmonary blood flow.
- Ventricular septal defect.
- Atrial septal defect.
- Patent ductus arteriosus.
- Increased pressure in the left atrium.
- Mitral valve defects.
- Myxoma or thrombus of the left atrium.
- Chronic left ventricular failure of any origin.
- Increased resistance in the pulmonary artery system.
- Hypoxic genesis (chronic obstructive pulmonary diseases, high-altitude hypoxia, hypoventilation syndrome).
- Obstructive genesis (recurrent pulmonary embolism, influence of pharmacological agents, primary pulmonary hypertension, diffuse connective tissue diseases, systemic vasculitis, veno-occlusive disease).
Symptoms pulmonary hypertension
The first clinical symptoms of pulmonary hypertension appear when the blood pressure in the pulmonary artery increases by 2 times or more compared to the norm.
The main subjective manifestations of pulmonary hypertension are practically the same in all etiological forms of this syndrome. Patients are concerned about:
- shortness of breath (the earliest and most common complaint of patients) initially during physical exertion, and later at rest;
- weakness, increased fatigue;
- fainting (caused by hypoxia of the brain, most typical for primary pulmonary hypertension);
- pain in the heart area of a constant nature (in 10-50% of patients, regardless of the etiology of pulmonary hypertension); caused by relative coronary insufficiency due to severe hypertrophy of the myocardium of the right ventricle;
- hemoptysis is a common symptom of pulmonary hypertension, especially with a significant increase in pressure in the pulmonary artery;
- hoarseness of voice (noted in 6-8% of patients and caused by compression of the left recurrent nerve by a significantly dilated pulmonary artery);
- pain in the liver area and swelling in the feet and shins (these symptoms appear with the development of pulmonary heart failure in patients with pulmonary hypertension).
Progressive dyspnea on exertion and easy fatigability are present in almost all cases. Dyspnea may be accompanied by atypical chest discomfort and dizziness or light-headedness on exertion. These symptoms of pulmonary hypertension are caused primarily by inadequate cardiac output. Raynaud's phenomenon occurs in approximately 10% of patients with primary pulmonary hypertension, of whom 99% are women. Hemoptysis is rare but can be fatal; dysphonia due to compression of the recurrent laryngeal nerve by an enlarged pulmonary artery (Ortner's syndrome) is also rare.
In advanced cases, symptoms of pulmonary hypertension may include right ventricular heave, diffuse second heart sound (S2) with accentuated pulmonary component of S (P), pulmonary ejection click, right ventricular third heart sound (S3), and jugular venous distension. Liver congestion and peripheral edema are common in advanced stages.
Portopulmonary hypertension
Portopulmonary hypertension is severe pulmonary arterial hypertension with portal hypertension in patients without secondary causes.
Pulmonary hypertension occurs in patients with a variety of conditions that result in portal hypertension with or without cirrhosis. Portopulmonary hypertension is less common than hepatopulmonary syndrome in patients with chronic liver disease (3.5-12%).
The first symptoms are shortness of breath and fatigue, and chest pain and hemoptysis may also occur. Patients have physical findings and ECG changes characteristic of pulmonary hypertension; signs of cor pulmonale (jugular venous pulsation, edema) may develop. Tricuspid regurgitation is common. The diagnosis is suspected on the basis of echocardiography and confirmed by right heart catheterization.
Treatment - therapy of primary pulmonary hypertension, excluding hepatotoxic drugs. In some patients, vasodilator therapy is effective. The outcome is determined by the underlying liver pathology. Portopulmonary hypertension is a relative contraindication to liver transplantation due to the increased risk of complications and mortality. After transplantation, some patients with moderate pulmonary hypertension experience reversal of the pathology.
Where does it hurt?
What's bothering you?
Diagnostics pulmonary hypertension
An objective examination reveals cyanosis, and with prolonged pulmonary hypertension, the distal phalanges of the fingers acquire the shape of “drumsticks,” and the nails take on the appearance of “watch glasses.”
Auscultation of the heart reveals characteristic signs of pulmonary hypertension - accentuation (often splitting) of the second tone over a.pulmonalis; systolic murmur over the region of the xiphoid process, increasing on inspiration (Rivero-Corvallo symptom) - a sign of relative insufficiency of the tricuspid valve, formed in connection with pronounced hypertrophy of the myocardium of the right ventricle; in later stages of pulmonary hypertension, diastolic murmur can be detected in the second intercostal space on the left (over a.pulmonalis), caused by relative insufficiency of the pulmonary artery valve with its significant expansion (Graham-Still murmur).
Percussion of the heart usually does not reveal symptoms pathognomonic of pulmonary hypertension. Rarely, it is possible to detect an expansion of the border of vascular dullness in the 2nd intercostal space on the left (due to expansion of the pulmonary artery) and a shift of the right border of the heart outward from the right parasternal line due to hypertrophy of the myocardium of the right ventricle.
Pathognomonic for pulmonary hypertension are: hypertrophy of the right ventricle and right atrium, as well as signs indicating an increase in pressure in the pulmonary artery.
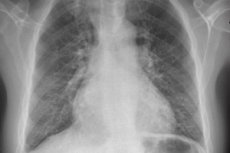
To detect these symptoms, the following are used: chest X-ray, ECG, echocardiography, right heart catheterization with pressure measurement in the right atrium, right ventricle, and pulmonary artery trunk. When performing right heart catheterization, it is also advisable to determine pulmonary capillary pressure or pulmonary artery wedge pressure, which reflects the pressure level in the left atrium. Pulmonary artery wedge pressure increases in patients with heart disease and left ventricular failure.
To identify the causes of pulmonary hypertension, it is often necessary to use other methods of examination, such as X-ray and computed tomography of the lungs, ventilation-perfusion radionuclide scintigraphy of the lungs, angiopulmonography. The use of these methods allows us to determine the pathology of the parenchyma and vascular system of the lungs. In some cases, it is necessary to resort to lung biopsy (to diagnose diffuse interstitial lung diseases, pulmonary veno-occlusive disease, pulmonary capillary granulomatosis, etc.).
In the clinical picture of pulmonary heart disease, hypertensive crises in the pulmonary artery system may be observed. The main clinical manifestations of the crisis are:
- sudden suffocation (most often occurs in the evening or at night);
- severe cough, sometimes with sputum mixed with blood;
- orthopnea;
- severe general cyanosis;
- arousal is possible;
- pulse is frequent and weak;
- pronounced pulsation of the a.pulmonalis in the 2nd intercostal space;
- bulging of the conus a.pulmonalis (on percussion it is manifested by the expansion of vascular dullness in the 2nd intercostal space on the left);
- pulsation of the right ventricle in the epigastrium;
- accent of the second tone on a.pulmonalis;
- swelling and pulsation of the jugular veins;
- the appearance of vegetative reactions in the form of urina spastica (the release of a large amount of light urine with low density), involuntary defecation after the end of the crisis;
- the appearance of the Plesh reflex (hepatojugular reflex).
The diagnosis of primary pulmonary hypertension is suspected in patients with significant dyspnea on exertion in the absence of a history of other diseases that could cause pulmonary hypertension.
Patients initially undergo chest x-ray, spirometry, and ECG to identify more common causes of dyspnea, followed by Doppler echocardiography to measure right ventricular and pulmonary artery pressures and to identify possible anatomical abnormalities causing secondary pulmonary hypertension.
The most common radiographic finding in primary pulmonary hypertension is widened hilars with marked peripheral narrowing (‘clipped’). Spirometry and lung volumes may be normal or show mild restriction, but the diffusing capacity for carbon monoxide (DL) is usually decreased. Common ECG changes include right axis deviation, R > S in V; SQ T waves, and peaked P waves.
Additional investigations are performed to diagnose secondary causes that are not clinically apparent. These include ventilation-perfusion scanning to detect thromboembolic disease; pulmonary function tests to identify obstructive or restrictive lung diseases; and serologic tests to confirm or exclude rheumatic diseases. Chronic thromboembolic pulmonary arterial hypertension is suggested by CT or lung scanning and is diagnosed by arteriography. Other investigations, such as HIV testing, liver function tests, and polysomnography, are performed in appropriate clinical situations.
If the initial evaluation does not reveal any conditions associated with secondary pulmonary hypertension, pulmonary artery catheterization should be performed to measure right heart and pulmonary artery pressures, pulmonary capillary wedge pressure, and cardiac output. Right atrial septal defect should be ruled out by measuring the O 2 saturation. Primary pulmonary hypertension is defined as a mean pulmonary artery pressure greater than 25 mm Hg in the absence of possible causes. However, most patients with primary pulmonary hypertension have significantly higher pressures (eg, 60 mm Hg). Vasodilators (eg, inhaled nitric oxide, intravenous epoprostenol, adenosine) are often used during the procedure; the decrease in right ventricular pressure in response to these drugs helps guide drug selection. Biopsy was once widely used but is no longer recommended because of its high morbidity and mortality.
If a patient is diagnosed with primary pulmonary hypertension, the family history is examined to identify possible genetic transmission, which is indicated by cases of premature death of relatively healthy people in the family. In familial primary pulmonary hypertension, genetic counseling is necessary to inform family members of the risk of the disease (approximately 20%) and to recommend them to undergo screening (echocardiography). In the future, testing for mutations in the BMPR2 gene in familial primary pulmonary hypertension may be of value.
What do need to examine?
What tests are needed?
Who to contact?
Treatment pulmonary hypertension
Treatment of secondary pulmonary hypertension is aimed at treating the underlying pathology. Patients with severe pulmonary arterial hypertension due to chronic thromboembolism should undergo pulmonary thromboendarterectomy. This is a more complex operation than emergency surgical embolectomy. In the extrapulmonary circulation, the organized vascularized thrombus is excised along the pulmonary trunk. This procedure cures pulmonary arterial hypertension in a significant percentage of cases and restores extrapulmonary function; in specialized centers, the operative mortality is less than 10%.
Treatment of primary pulmonary hypertension is evolving rapidly. It begins with oral calcium channel blockers, which can reduce pulmonary artery pressure or pulmonary vascular resistance in approximately 10% to 15% of patients. There is no difference in efficacy between the different types of calcium channel blockers, although most experts avoid verapamil because of its negative inotropic effects. Response to this therapy is a favorable prognostic sign, and patients should continue this treatment. If there is no response, other drugs are started.
Intravenous epoprostenol (a prostacyclin analogue) improves function and prolongs survival even in patients resistant to vasodilators at the time of catheterization. Disadvantages of treatment include the need for an indwelling central catheter and significant adverse effects, including flushing, diarrhoea, and bacteremia due to long-term central catheter indwelling. Alternative agents—inhaled (iloprost), oral (beraprost), and subcutaneous (treprostinil) prostacyclin analogues—are under study.
The oral endothelin receptor antagonist bosentan is also effective in some patients, typically those with milder disease and not responsive to vasodilators. Oral sildenafil and L-arginine are also under investigation.
Forecast
Lung transplantation offers the only hope for a cure, but carries a high risk of complications due to rejection problems and infection. The five-year survival rate is 60% due to bronchiolitis obliterans. Lung transplantation is reserved for patients with New York Heart Association stage IV heart failure (defined as shortness of breath with minimal activity, requiring confinement to bed or chair) who have not responded to prostacyclin analogs.
Many patients require additional medications to treat heart failure, including diuretics, and they must also receive warfarin to prevent thromboembolism.
The median survival of patients without treatment is 2.5 years. The cause is usually sudden death due to right ventricular failure. Five-year survival with epoprostenol is 54%, while in the minority of patients who respond to calcium channel blockers, it exceeds 90%.
Pulmonary hypertension has a poor prognosis if symptoms such as low cardiac output, higher pulmonary artery and right atrial pressures, lack of response to vasodilators, heart failure, hypoxemia, and deterioration of overall functional status are present.