Medical expert of the article

New publications
Hamartoma
Last reviewed: 29.06.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
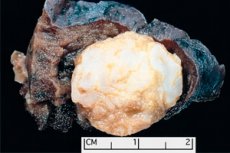
Tumor-like formation localized in any anatomical region resulting from abnormal growth of benign tissue, in medicine is defined as hamartoma (from Greek hamartia - error, defect). [1]
Epidemiology
Statistically, hamartomas account for 1.2% of benign neoplasms. The prevalence of pulmonary hamartomas is estimated to be approximately 0.25% of the general population and accounts for up to 8% of all pulmonary neoplasms. Most pulmonary hamartomas are diagnosed incidentally in patients 40 to 70 years of age, but are very rare in pediatric practice.
In general, most hamartomas are diagnosed in men, although in the kidney they are more common in women and are identified in middle age.
About 5% of benign breast tumors are hamartomas, and they most commonly affect women over the age of 35.
80-90% of hamartomatous lesions of the brain and more than 50% of hamartomas of the heart are associated with tuberous sclerosis.
Causes of the hamartomas
Gamartomas belong to congenital malformations and are formations of benign character, which are formed from mesenchymal tissues originating from germinal sheets. And the causes of their occurrence are associated with uncontrolled cell division of cytologically normal tissues (connective, smooth muscle, fat or cartilage), characteristic of a given anatomical location, and their focal overgrowth during embryogenesis of almost any organ or anatomical structure.
The occurrence of multiple hamartomas in the same patient is often referred to as hamartomatosis or pleiotropic hamartoma.
These tumors can occur sporadically or in the presence of certain autosomal dominant inherited diseases as well as genetically determined syndromes.
In many cases, hamartomas form when a rare genetic disease of multisystemic nature - tuberous sclerosis - manifests shortly after birth, or in Recklinghausen's familial disease - neurofibromatosis type 1. [2]
Risk factors
The main risk factors for hamartoma formation include the presence of so-called genetic syndromes of hamartomatous polyposis in the patients' history, including:
- Multiple hamartoma syndrome - Cowden syndrome, in which multiple hamartomas of ecto-, ento- and mesodermal origin are formed, gastrointestinal polyposis and mucocutaneous manifestations are observed;
- Peutz-Jeghers-Turen syndrome (characterized by the development of benign hamartomatous polyps in the gastrointestinal tract);
- Proteus syndrome;
- Weil syndrome - juvenile polyposis of the colon;
- Bannayan-Riley-Ruvalcaba syndrome, which, like Cowden syndrome, produces multiple hamartomas (hamartomatous polyps) of the intestine;
- Carney-Stratakis syndrome and Carney complex.
In addition, hamartomas form in patients with hereditary Watson syndrome and in cases of sporadically occurring or congenital Pallister-Hall syndrome with hypothalamic hamartoma and polydactyly.
Pathogenesis
The mechanism of increased proliferation of germinal tissues with the formation of tumor-like malformations in various organs is explained by chromosomal aberrations and gene mutations that can occur spontaneously or be inherited.
In tuberous sclerosis, mutations in the TSC1 or TSC2 genes - tumor suppressors that prevent and inhibit excessive proliferation - too rapid or uncontrolled cell growth and division - have been identified. And in neurofibromatosis type 1 and Watson syndrome - germline mutations of the mitochondrial tumor suppressor gene NF1.
In hamartoma tumor syndrome, which combines Cowden, Protea, Bannayan-Riley-Ruvalcaba, and juvenile polyposis syndromes, pathogenesis is associated with mutation of the PTEN gene, which encodes an enzyme involved in the regulation of proliferation and is considered a tumor suppressor gene.
Mutations in the STK11 gene encoding the structure and function of one of the transmembrane serine enzymes, which reduces its ability to restrain cell division, lead to Peutz-Jeghers-Turen syndrome, with the development of intestinal polyps and pigmented skin lesions. A mutation in the GLI3 gene, a transcription factor involved in intrauterine tissue formation, has been identified in Pallister-Hall syndrome.
Thus, uncontrolled cell growth due to gene mutations leads to hamartoma formation.
Symptoms of the hamartomas
Depending on the localization of hamartomas, their types are distinguished, and each of them has its own structure and symptomatology.
Hamartoma of the lung
Pulmonary hamartoma can form in any lobe and peripheral parts of the lungs and consists of normal tissues present in the lungs: adipose, epithelial, fibrous and cartilaginous. In 80% of cases, the chondroid component (hyaline cartilage cells) predominates with the inclusion of adipocytes - adipose tissue cells and airway epithelial cells. [3]
The earlier names: chondroid hamartoma, mesenchymoma, chondromatous hamartoma, or hamartochondroma are not currently recommended by WHO.
Mesenchymal cystic hamartoma of the lung, on the other hand, is less common and is associated with Cowden syndrome in most patients.
Hamartomatous lesion of the lung may not manifest itself, but may cause symptoms in the form of chronic cough (often with hemoptysis), wheezing when breathing and difficulty breathing. [4]
A hamartoma of the heart
Benign primary cardiac tumors in adults include mature myocyte hamartoma, and in infants and children with tuberous sclerosis, rhabdomyoma, that is, myocardial hamartoma of the ventricles or interventricular septum. [5]
Mature cardiomyocyte hamartoma develops in the ventricular wall (and rarely in the atria) and may appear as multiple lesions that are dense masses closely associated with the underlying myocardium. The tumor may cause symptoms of heart failure: chest pain, palpitations and arrhythmias, heart murmurs, edema, dyspnea, cyanosis.
Cardiac rhabdomyomas, most of which are diagnosed within the first year of life, are composed of cardiac muscle tissue formed by embryonic myoblasts and have the appearance of solid focal masses without a capsule.
Typically, these hamartomas present asymptomatically and spontaneously regress before the age of 4 years.
Hamartomatous lesions are also considered by some experts to be associated with the Carney complex myxoma of the heart. [6]
Gamartoma of the gastrointestinal tract
Gastric hamartoma is a mesenchymal mass in the form of an epithelial hyperplastic polyp of the stomach, Peutz-Jeghers polyp and a rare myoepithelial hamartoma - with hypertrophied smooth muscle bundles. Other names for this hamartoma include myoglandular hamartoma, adenomyomatous hamartoma, and gastric adenomyoma. Typical clinical manifestations include dyspepsia, epigastric pain, and upper GI bleeding. [7], [8]
More information in the material - gastric Polyposis
An intestinal hamartoma is a hamartomatous or hyperplastic polyp of the large intestine, diagnosed as an adenomatous or tubular adenoma. When the hamartoma is localized in the Brunner's gland of the duodenum, symptoms are manifested by pain in the epigastric region; nausea, vomiting, and flatulence (indicating intestinal obstruction); and, if of considerable size, gastrointestinal bleeding. In cases of myoepithelial hamartoma of the ileum, patients complain of abdominal pain, have decreased body weight, and develop chronic anemia. [9], [10]
Also read - rectal Polyps
Retrorectal hamartoma is a cystic hamartoma or multichambered cyst of the retrorectal space (the loose connective tissue between the rectum and its own fascia) that most commonly occurs in middle-aged women. It has the appearance of a cyst bulging out of the posterior wall of the rectum, which is lined with epithelium and contains chaotically arranged smooth muscle fibers. This hamartoma presents with lower abdominal pain and recurrent constipation. [11], [12]
Hamartomas of the liver and spleen
Multiple biliary hamartoma of the liver is a hamartoma of the interdollic intrahepatic bile ducts associated with malformations of their development during the embryonic period. This hamartoma (single or multiple) consists of haphazardly dilated clusters of bile ducts and fibrocollagenous stroma. [13]
Biliary hamartomas are asymptomatic and are usually discovered incidentally (during radiologic examination or laparotomy). [14]
A rare and often incidentally detected primary neoplasm of benign character is a hamartoma of the spleen, which consists of elements of the red pulp of the spleen - in the form of a well-defined homogeneous mass of firm consistency. This malformation can be single or multiple; when squeezing the splenic parenchyma, there may be a feeling of discomfort and pain in the left subcostal area. [15], [16]
Renal hamartomas
The most common hamartoma of the kidney is diagnosed as angiomyolipoma of the kidney, as this benign tumor consists of mature adipose tissue with embedded smooth muscle fibers and blood vessels. It forms in tuberous sclerosis in 40-80% of cases. Increasing the size of hamartoma (more than 4-5 cm) leads to pain and the appearance of blood in the urine. [17], [18]
Hamartoma of the breast
The WHO-accepted diagnostic definitions of breast hamartoma are terms such as adenolipoma, chondrolipoma, and myoid hamartoma. Although often called fibroadenolipoma by mammologists, because the tumor formation contains cells of fibrous, glandular, and adipose tissue enclosed in a thin connective tissue capsule with distinct outlines. Focal calcifications may be observed on visualization. In this case, clinical manifestations are absent. [19], [20]
Also read - breast Tumors
Hamartomas of the brain
One third of patients with tuberous sclerosis have a brain hamartoma in the form of intracranial cortical outgrowths or tubercles in various lobes - at the border of gray and white matter - or subependymal nodules along the walls of the brain ventricles. Astrocytic hamartoma, a subependymal giant-cell astrocytoma with cortical disruption, dysmorphic neurons and large glial cells of the brain parenchyma (astrocytes), may also form. Symptoms of cerebral hamartomas include seizure attacks and mental retardation in children. [21], [22]
A rare malformation that occurs during embryogenesis and is present at birth is a hypothalamic hamartoma, which is a mass of heterotopic neurons and glial cells. As the child's brain grows, the tumor enlarges but does not spread to other cerebral regions. [23], [24]
If hypertrophied tissues are formed in the anterior part of the hypothalamus (tuber cinereum), where the pituitary gland attaches to it, malformation manifests symptoms of central premature sexual development (before 8-9 years of age): appearance of acne rashes, early development of mammary glands and early menarche in girls; early pubic hair and voice mutation in boys.
When hamartomas form in the posterior part of the hypothalamus, there may be abnormalities in the electrical activity of the brain, which in early infancy is manifested by seizures, and at a later stage (ages 4 to 7 years) by epilepsy with focal epileptic seizures with sudden laughter or with involuntary crying, atonic and tonic-clonic seizures, as well as seizures of aggression, memory and cognitive problems.
A pituitary hamartoma is a sporadically occurring benign pituitary adenoma.
Middle-aged adults with Cowden syndrome may have a rare tumor-like mass, a hamartoma of the cerebellum, diagnosed as dysplastic cerebellar gangliocytoma or Lhermitte-Duclos disease. Symptoms may be absent or manifest as headache, dizziness, impaired coordination of movements, and paralysis of individual cranial nerves.
Lymph node hamartoma
When the cells of smooth muscle and adipose tissue, as well as blood vessels and collagenous stroma of inguinal, retroperitoneal, submandibular and cervical lymph nodes overgrow, an angiomyomatous hamartoma of a lymph node or nodular angiomyomatous hamartoma is formed - with partial or complete replacement of its parenchyma. [25], [26]
A hamartoma of the skin
In the presence of tuberous sclerosis or neurofibromatosis, various hamartomas of the skin are observed, most often in the form of hypopigmented spots; coffee and milk spots; angiofibroma (on the cheeks, chin, nasolabial folds); shagreen spots of various localizations (which are connective tissue nevi); fibrous plaques on the forehead, scalp or neck.
A rare dermatologic manifestation of tuberous sclerosis (especially in men) is folliculocystic and collagen hamartoma, characterized by abundant collagen deposition in the dermis, concentric perifollicular fibrosis, and keratin-filled funnel-shaped subcutaneous cysts seen on histopathologic examination. [27]
To hamartomas consisting of melanocytes (cells that produce the pigment melanin), most experts also refer to various melanocytic neoplasms, in particular congenital melanocytic nevi, which represent an abnormality of embryogenesis.
In terms of etiology, hamartomas composed of vascular tissue are also hemangiomas of the skin.
Patients with Peutz-Jeghers-Thuren syndrome have hamartoma in the form of patchy pigmentation of the skin and mucous membranes - lentiginosis periorificialis
Cases of linear papular ectodermal-mesodermal hamartoma (Hamartoma moniliformis) show a linear flesh-colored papular rash on the head, neck, and upper chest.
And a sebocytic hamartoma is a hamartoma of the sebaceous glands, read more in the publication - sebaceous nevus.
Hamartoma of the eye
Pigmented hamartomatous lesions of the iris in neurofibromatosis type 1 and Watson syndrome - in the form of nodular clusters of dendritic melanocytes - are defined as iris hamartomas or Lisch nodules. They are transparent (usually not affecting vision) rounded dome-shaped yellow-brown papules that protrude above the surface of the iris.
And patients with juvenile angiofibroma of the nasopharynx and familial adenomatous polyposis often develop combined hamartoma of the retina and retinal pigment epithelium - in the form of a black spot on the central (macular) part of the retina. [28]
A hamartoma of the nose
Nasal hamartoma is defined by specialists as nasal chondromesenchymal hamartoma or nasal chondroma, due to benign proliferation of respiratory epithelium, submucosal glands and chondro-bone mesenchyme. Its clinical manifestations depend on the size and localization of the lesion and include: nasal congestion, difficulty in nasal breathing and breastfeeding in infants, clear watery nasal discharge, and nasal bleeding. A hamartoma may grow with the child and spread into the eye orbits, resulting in forward or backward displacement of the eyeball, strabismus, or oculomotor disturbances. [29]
A hamartoma in a child
All the above-mentioned hamartomatous lesions of various organs and anatomical structures are present in children with corresponding syndromes.
Newborns present with mesenchymal hamartoma of the chest wall or cartilaginous hamartoma of the rib, which are solid immobile masses resulting from focal overgrowth of normal skeletal elements with cartilaginous, vascular and mesenchymal elements. This hamartoma may cause respiratory failure and the development of respiratory distress syndrome. Mesenchymal hamartoma of the liver is the second most frequent benign liver tumor in children. This tumor-like formation (more often localized in the right lobe of the organ) consists of cells of mesenchymal stroma, hepatocytes and epithelial cells of bile duct lining. The clinical picture includes palpable mass in the abdominal cavity, anorexia and weight loss, and in case of significant size (up to 10 cm and more) the tumor covers extrahepatic bile ducts and inferior vena cava, which leads to jaundice and edema of lower extremities.
A hamartoma is a congenital mesoblastic nephroma (occurring in 1 in 200,000 infants) that may result in abdominal bloating in the newborn with a palpable mass of dense consistency in the right upper quadrant of the abdomen. Infants may also present with rapid shallow breathing.
Rare congenital anomalies include fibrous hamartoma of infancy, which occurs in children in the first two years of life and presents as a painless nodular mass in the subcutaneous tissues of the axilla, neck, shoulder and forearm, back and chest, thigh, foot, and external genitalia.
Eccrine angiomatous hamartoma in a child may be present at birth or manifest in early childhood. This benign tumor of hamartomatous nature usually has the appearance of bluish or brownish nodules and/or plaques that result from proliferation of eccrine sweat gland tissue and capillaries in the middle and deep layers of the dermis. This hamartoma may cause localized hyperhidrosis and increased hair growth.
Complications and consequences
It is generally agreed that hamartomas rarely recur or transform into malignant tumors. They often show little or no symptoms and sometimes even disappear over time. But in more severe cases and depending on the site of formation, these malformations can have serious complications and consequences.
First of all, a hamartoma can grow to such a size that it begins to press on surrounding tissues and organs, disrupting their functions.
Cardiac hamartoma in children can lead to persistent heart rhythm abnormalities, valve defects, and impaired intracardiac blood flow with subsequent congestive heart failure.
Complications of hamartomatous polyps of the GI tract are gastrointestinal bleeding, obstruction and intestinal intussusception (with possible fatal outcome). And a large renal hamartoma can provoke rupture of the kidney.
A hamartoma in the brain can cause obstructive hydrocephalus syndrome.
In hypothalamic and pituitary hamartomas, the production of somatotropic hormone (growth hormone) may be impaired, leading to the development of hypophyseal nanism (hypopituitarism) in children. Hypothalamic hamartomas in children can also lead to drug-resistant epilepsy.
Complications of retinal pigment epithelium hamartoma are fraught with retinal and/or optic nerve dysfunction, macular edema, neovascularization of the choroid, and retinal detachment.
Diagnostics of the hamartomas
An important part of the diagnosis of hamartomas and related syndromes is the collection of anamnesis, including family history.
Laboratory tests include blood tests: general clinical; serum electrolytes; lymphocyte profile; calcium, potassium, phosphate and urea levels; and liver function tests. If possible, a fine-needle aspiration puncture biopsy of the mass is performed, since histologic examination is crucial in the diagnosis and choice of treatment tactics.
Instrumental diagnostics provides visualization of hamartomatous tumor-like formation and identification of its exact localization, for which X-ray, angiography, electroencephalography (EEG), ultrasound (sonography), CT (computed tomography), PET (positron emission tomography), MRI (magnetic resonance imaging) are used.
Differential diagnosis
In any abnormal masses, differential diagnosis is very important. Thus, tuberculoma and hamartoma are differentiated; pulmonary hamartoma and primary lung cancer, bronchogenic carcinoid, metastatic disease. Brain hamartoma should be distinguished from craniopharyngioma and hypothalamic-chiasmatic glioma. And the differential diagnosis of hamartoma as congenital mesoblastic nephroma includes Wilms tumor (malignant nephroblastoma), clear cell sarcoma of the kidney and ossifying kidney tumor in infants.
Who to contact?
Treatment of the hamartomas
If the hamartoma is asymptomatic and is discovered accidentally, no treatment is required, but it is necessary to monitor its "behavior" and the patient's condition. In other cases, therapy is aimed at reducing the intensity of symptoms and preventing complications. For example, in hypothalamic hamartoma with symptoms of premature puberty, certain medications that inhibit the release of certain hormones are prescribed. Cardiac medications are used to treat symptoms of heart failure in patients with cardiac hamartomas.
Surgical removal of hamartomas is indicated to confirm the diagnosis and in cases of medically uncorrectable intense symptoms.
For example, lung hamartomas may be resected by wedge resection and, in severe cases, by removal of a lobe of the lung (lobectomy). A breast hamartoma may also be excised, and if it is large, a partial or complete mastectomy may be required.
Stereotactic radiofrequency thermoablation or laser ablation can be used to remove hamartomatous polyps. Radiosurgery with highly focused gamma rays - gamma knife for hypothalamic hamartomas or astrocytic hamartomas - is also used.
Prevention
The only method of preventing the development of hamartomas can be considered genetic screening of the child's future parents.
Forecast
The overall prognosis of this congenital anomaly depends on the localization and size of the neoplasm, as well as on comorbidities and the general health of the patient.