Medical expert of the article

New publications
Neuroblastoma in children: causes, diagnosis, treatment
Last reviewed: 04.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
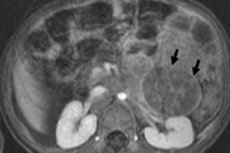
In pediatric oncology, one of the most common extracranial neoplasms is neuroblastoma in children, which is an embryonic malignant tumor of neural crest neuroblasts, that is, embryonic (immature) nerve cells of the sympathetic nervous system.
Epidemiology
According to statistics from the International Neuroblastoma Risk Group (INRG), neuroblastoma accounts for about 8% of all oncological diseases in children worldwide and is third in prevalence, after leukemia and brain tumors.
According to other data, neuroblastoma accounts for about 28% of all cancers in infants. More than a third of cases of neuroblastoma are diagnosed in children under one year of age; the average age of diagnosis is 19-22 months. More than 90% of diagnosed cases occur in children aged two to five years (with a predominance of boys); the peak incidence is observed at the age of two to three years, and cases in children over five years of age account for less than 10%.
Causes neuroblastomas
In studying the causes of neuroblastoma, researchers have concluded that this tumor in children occurs due to sporadic genetic mutations during embryogenesis or early postnatal development. But what causes these gene changes is unknown, since no influence of teratogenic environmental factors has been identified.
These tumors can occur anywhere, including the mediastinum, neck, abdomen, adrenal glands, kidneys, spine, and pelvis.
In rare cases, neuroblastoma in infants may be associated with an inherited mutation. In particular, a mutation in the gene of the membrane protein CD246 on chromosome 2 - the enzyme tyrosine kinase ALK, which ensures intercellular communications and plays an important role in the functioning of the nervous system; in the gene of the protein PHOX2B (on chromosome 4), which is involved in the maturation of nerve cells.
Neuroblastoma may also be associated with childhood neurofibromatosis type 1,Beckwith-Wiedemann syndrome, and hyperinsulinemic hypoglycemia (nesidioblastosis pancreatitis).
Risk factors
Today, heredity is recognized as risk factors for the development of neuroblastoma in children - the presence of this tumor in the family history, as well as congenital anomalies associated with gene mutations during intrauterine development. This is especially true for cases of the development of several neoplasms in different organs.
None of the exogenous factors that increase the risk of this tumor have been identified by researchers.
Pathogenesis
The mechanism of development of neuroblastomas is caused by disturbances in differentiation and maturation of neural crest cells – bilateral cell lines that form at the edges of the neural tube from the ectodermal germ layer of the human embryo. These cells migrate (move) and differentiate into many types of cells: sensory and autonomic neurons, neuroendocrine cells and cells of the adrenal medulla, cells of the craniofacial cartilage and bones, as well as pigment cells.
In neuroblastoma, the migrated neuroblasts do not mature, but continue to grow and divide, forming a tumor. And the pathogenesis of its formation is associated with the following gene mutations:
- with duplication of part of the chromosome sequence or duplication of segments of the LMO1 gene on chromosome 11, encoding the RBTN1 protein in the neural crest cells of the embryo;
- with a change in the copy number of the NBPF10 gene on chromosome 1q21.1, encoding the DUF1220 protein, which controls the proliferation of human neural stem cells. These disorders lead to either a duplication of this chromosome or to its deletion - the absence of part of the DNA;
- with changes in the tumor suppressor gene ATRX (on chromosome Xq21.1);
- with the presence of additional copies (amplification) of the N-Myc transcription factor gene on chromosome 2, which codes for one of the transcription factors (DNA-binding protein) that regulates the activity of other genes and controls the proliferation of precursor cells during the formation of proteins for the formation of tissues and organs of the fetus. Amplification of this gene turns it into an oncogene, which provokes a disruption of the cell cycle, increased cell proliferation and tumor formation.
Symptoms neuroblastomas
The first signs of neuroblastoma are nonspecific and may include loss of appetite (and weight loss), fatigue when feeding, fever, and joint pain.
Clinical symptoms depend on the location of the primary tumor and the presence of metastases (which occur in 60-73% of cases).
Very often, primary neuroblastoma is localized in the adrenal medulla, which has a similar origin to nerve cells. In children under one year of age, adrenal neuroblastoma is diagnosed in 35-40% of cases. Its symptoms include abdominal pain, fever, weight loss, bone pain, anemia or concomitant Pepper's syndrome: diffuse liver damage with severe hepatomegaly and respiratory distress syndrome.
Retroperitoneal neuroblastoma or retroperitoneal neuroblastoma in children, as it grows, begins to press on the bladder or intestines, which can cause problems with urination or defecation, swelling of the legs (in boys, the scrotum swells).
Neuroblastoma of the mediastinum in children (mediastinal neuroblastoma) often presses on the superior vena cava, and this can cause swelling of the face, neck, arms, and upper chest (with the skin becoming bluish-red, with subcutaneous nodules). Coughing and wheezing, breathing problems (shortness of breath) or swallowing problems (dysphagia) appear; enlarged lymph nodes are noted in the neck, above the collarbone, and in the armpits.
The spread of tumor cells to the bone marrow leads to anemia, thrombocytopenia and leukopenia with a tendency to bleeding.
And with metastases in the periorbital area, dark circles or bruises appear around the eyes. Such a tumor can also cause headaches and dizziness, exophthalmia (bulging of the eyeballs), and due to compression of the nerve endings - drooping eyelids (ptosis) and a decrease in the size of the pupils (miosis).
Abdominal neuroblastoma or neuroblastoma of the abdominal cavity in children leads to the formation of palpable seals in the abdomen, its distension, loss of appetite, constipation, and increased blood pressure. A tumor pressing on the spinal cord or nerve root can lead to numbness and weakness of the limbs, inability to stand, crawl, or walk. If bones are affected, bone pain may occur.
In case of stage 3-4 tumor in the abdominal cavity with lymph node damage, tumor cells can enter the renal parenchyma, and then extensive neuroblastoma of the kidney in children develops, leading to disruption of its functions.
Stages
- Stage 1 neuroblastoma is a primary tumor that is localized and isolated to one area of the body; lymph nodes on both sides are not affected.
- Neuroblastoma stage 2. In stage 2A, the primary tumor is confined to one area but is large; bilateral lymph nodes are not involved. In stage 2B, the lymph nodes on the side of the body where the tumor is located are positive for metastases.
- Neuroblastoma stage 3: the primary tumor crosses the spinal cord or midline of the body, unilateral or bilateral metastases are found in the lymph nodes.
- Neuroblastoma stage 4: the tumor has spread to distant lymph nodes, bone marrow, bones, liver or other organs. And stage 4S is determined in children under one year old with a localized primary tumor, with dissemination to the skin, liver or bone marrow.
International Neuroblastoma Risk Staging System (INRGSS)
INRGSS uses imaging-defined risk factors (IDRFs), which are factors seen on imaging tests that may mean a tumour will be more difficult to remove.
INRGSS divides neuroblastomas into 4 stages:
- L1: The tumor has not spread from where it started and has not grown into vital structures. It is limited to one part of the body, such as the neck, chest, or abdomen.
- L2: The tumor has not spread (metastasized) far from where it started (for example, it may have grown from the left side of the abdomen to the left side of the chest), but it has at least one IDRF.
- M: The tumor has metastasized to a distant part of the body (except for tumors at the MS stage).
- MS: Metastatic disease in children under 18 months of age, in which cancer has spread only to the skin, liver, and/or bone marrow.
Complications and consequences
Neuroblastoma is characterized by complications and consequences such as:
- spread (metastasis) to the lymph nodes, bone marrow, liver, skin and bones;
- spinal cord compression (which can cause pain and lead to paralysis);
- development of paraneoplastic syndrome (due to the action of certain chemicals secreted by the tumor, as well as the antigen disialoganglioside GD2 expressed by its cells), which is manifested by rapid involuntary eye movements, impaired coordination, muscle cramps, and diarrhea;
- relapses after completion of primary therapy (as clinical practice shows, high-risk neuroblastomas have a relapse in 50% of cases).
Diagnostics neuroblastomas
Diagnosis of suspected neuroblastoma in a child requires examination, laboratory tests and imaging.
Blood and urine tests are taken for catecholamines (norepinephrine and dopamine) and homovanillic or vanillylmandelic acids (formed during the metabolism of these hormones); a blood test for neurospecific enolase, an enzyme-linked immunosorbent assay (ELISA) of the blood serum, and a bone marrow analysis (a sample of which is taken by aspiration puncture). A DNA test is performed to determine mutations, and a biopsy is performed for cytomorphological examination of the tumor tissue.
After the biopsy samples are taken, they are sent to a lab where they are looked at under a microscope by a pathologist (a doctor who has special training in identifying cancer cells). Special lab tests are also often run on the samples to show whether the tumor is neuroblastoma.
If it's neuroblastoma, lab tests can also help determine how quickly the tumor might grow or spread, as well as what treatments might work best.
Instrumental diagnostics visualizes the neoplasm using ultrasound, X-ray, MRI or CT, PET with the introduction of 18F-fluorodeoxyglucose or MIBG scanning - scintigraphy with metaiodobenzylguanidine. [ 1 ]
Differential diagnosis
Differential diagnosis includes benign ganglioneuroma, ganglioneuroblastoma, rhabdomyosarcoma, nephroblastoma.
Who to contact?
Treatment neuroblastomas
In neuroblastoma, treatment depends on the patient's risk group (stage of the tumor process), localization of the tumor, genomic features of tumor cells and the child's age. And it may include monitoring, surgery, chemotherapy, radiation therapy, immunotherapy, and hematopoietic stem cell transplantation.
Neoadjuvant or adjuvant (pre- or postoperative) chemotherapy for neuroblastoma in children, like any chemotherapy for cancer, is given in courses: the drug is administered for several days in a row, followed by a break for the body to recover. Cycles are usually repeated every three to four weeks.
The following medications (and their combinations) are used: Cyclophosphamide, Cisplatin or Carboplatin, Doxorubicin (Adriamycin), Vincristine, Etoposide.
Common side effects of chemotherapy drugs include hair loss, loss of appetite, fatigue, nausea and vomiting, mouth ulcers, diarrhea, or constipation. Chemotherapy can damage bone marrow and cause a decrease in blood cell count.
Targeted immunotherapy (aimed at the tumor antigen GD2) uses drugs from the group of monoclonal antibodies (anti-GD2 MAb) Dinutuximab (Unituxin) and Naxitamab. They are administered intravenously by prolonged infusion, in combination with granulocyte-macrophage colony-stimulating factor (cytokine GM-CSF) and interleukin-2.
Side effects of these drugs include pain (often very severe), decreased blood pressure, increased heart rate, shortness of breath (with possible swelling of the airways), increased temperature, nausea, vomiting and diarrhea, changes in the cellular and mineral composition of the blood.
To reduce the risk of cancer recurrence after high-dose chemotherapy and stem cell transplantation, children with high-risk neuroblastoma are treated with systemic retinoids, 13-cis-retinoic acid (Isotretinoin). [ 2 ]
Surgical treatment of neuroblastoma – tumor removal, for example, open adrenalectomy or laparoscopic resection of adrenal neuroblastoma; lymphectomy (removal of affected lymph nodes), etc. [ 3 ]
For high-risk neuroblastoma, radiation therapy may be used.[ 4 ]
Prevention
Given the causes of neuroblastoma in children, the only preventive measure may be genetic counseling when planning a pregnancy. But it should be borne in mind that this tumor is associated with inherited mutations only in 1-2% of cases.
Forecast
Infantile neuroblastoma has the ability to spontaneously regress.
Prognostic markers
- High-risk tumors, as well as neuroblastoma in children of all age groups and all stages (except stage 4S) – with increased expression of the N-MYC gene and amplification of the N-Myc oncogene – have an unfavorable prognosis that affects life expectancy.
- Having tumor cells that are missing certain parts of chromosomes 1 or 11 (known as 1p or 11q deletions) has a worse prognosis. Having an extra part of chromosome 17 (17q gain) is also associated with a worse prognosis.
- Neuroblastoma cells with a large amount of DNA have a better prognosis, especially for children under 2 years of age.
- Neuroblastomas that have more neurotrophin receptors, especially the nerve growth factor receptor TrkA, have a better prognosis.
Survival by Childhood Oncology Group (COG) risk group
- Low-risk group: Children in the low-risk group have a 5-year survival rate greater than 95%.
- Intermediate risk group: Children in the intermediate risk group have a 5-year survival rate of 90% to 95%.
- High-risk group: Children in the high-risk group have a 5-year survival rate of about 50%.
About 15% of childhood cancer deaths are due to neuroblastoma. The chances of long-term survival for this high-risk malignancy are no more than 40%. The overall five-year survival rate is 67-74%, 43% in the one-to-four age group, and more than 80% for neuroblastoma diagnosed in the first year of life.
Использованная литература