Medical expert of the article

New publications
Prions - causative agents of prion diseases
Last reviewed: 06.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
Slow viral infections are characterized by special criteria:
- an unusually long incubation period (months, years);
- a specific lesion of organs and tissues, primarily the central nervous system;
- slow, steady progression of the disease;
- inevitable fatal outcome.

Some pathogens that cause acute viral infections can also cause slow viral infections. For example, the measles virus sometimes causes SSPE, and the rubella virus causes progressive congenital rubella and rubella panencephalitis.
A typical slow viral infection of animals is caused by the visna/madi virus, which is a retrovirus. It is the causative agent of slow viral infection and progressive pneumonia in sheep. The white matter of the brain is destroyed, paralysis develops (visna - wasting away); chronic inflammation of the lungs and spleen occurs.
Diseases similar in their features to slow viral infections are caused by prions - the causative agents of prion infections. Prion diseases are a group of progressive disorders of the central nervous system of humans and animals. In humans, the function of the central nervous system is impaired, personality changes occur, and movement disorders occur. The symptoms of the disease usually last from several months to several years, ending in death. Previously, prion infections were considered together with the so-called causative agents of slow viral infections.
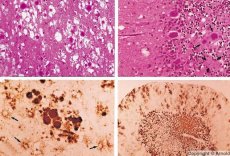
Some agents that cause prion diseases accumulate first in lymphoid tissues. Prions, getting into the brain, accumulate in large quantities, causing amyloidosis (extracellular dysproteinosis, characterized by the deposition of amyloid with the development of atrophy and sclerosis of the tissue) and astrocytosis (proliferation of astrocytic neuroglia, hyperproduction of glial fibers). Fibrils, aggregates of protein or amyloid and spongiform changes in the brain (transmissible spongiform encephalopathies) are formed. As a result, behavior changes, coordination of movements is impaired, exhaustion with a fatal outcome develops. Immunity is not formed. Prion diseases are conformational diseases that develop as a result of incorrect folding (violation of the correct conformation) of cellular protein necessary for the normal functioning of the body. The routes of prion transmission are varied:
- alimentary route - infected products of animal origin, food additives from raw bovine organs, etc.:
- transmission through blood transfusion, administration of drugs of animal origin, organ and tissue transplantation, use of infected surgical and dental instruments;
- transmission through immunobiological preparations (infection of 1500 sheep with PrP''' by brain formol vaccine from sick sheep is known).
Pathological prions, having entered the intestine, are transported into the blood and lymph. After peripheral replication in the spleen, appendix, tonsils and other lymphoid tissues, they are transferred to the brain through the peripheral nerves (neuroinvasion). Direct penetration of prions into the brain through the blood-brain barrier is possible. Previously, it was believed that the central nervous system is the only tissue in which pathological prions accumulate, but studies have appeared that have changed this hypothesis. It turned out that the accumulation of prions in the spleen is associated with the increase and functioning of follicular dendritic cells.
 [
[ Properties of prions
The normal cellular isoform of the prion protein with a molecular weight of 33-35 kDa is determined by the prion protein gene (the prion gene - PrNP is located on the 20th human chromosome). The normal gene appears on the cell surface (anchored in the membrane by the glycoprotein of the molecule), sensitive to protease. It regulates the transmission of nerve impulses, daily cycles, oxidation processes, participates in copper metabolism in the central nervous system and in the regulation of bone marrow stem cell division. In addition, the prion gene is found in the spleen, lymph nodes, skin, gastrointestinal tract and follicular dendritic cells.
Proliferation of pathological prions
The transformation of prions into altered forms occurs when the kinetically controlled equilibrium between them is disrupted. The process is enhanced by an increase in the amount of pathological (PrP) or exogenous prion. PrP is a normal protein anchored in the cell membrane. PrP' is a globular hydrophobic protein that forms aggregates with itself and PrP'' on the cell surface: as a result, PrP' is transformed into PrP'' and then the cycle continues. The pathological form of PrP''' accumulates in neurons, giving the cell a spongy appearance.
Kuru
Prion disease, previously common among the Papuans (meaning trembling or shaking) in the eastern part of the island of New Guinea. The infectious properties of the disease were proven by K. Gajdusek. The pathogen is transmitted by food as a result of ritual cannibalism - eating the insufficiently cooked, prion-infected brain of dead relatives. As a result of damage to the central nervous system, movement and gait are impaired, chills and euphoria ("laughing death") appear. The incubation period lasts 5-30 years. The patient dies after a year.
Creutzfeldt-Jakob disease
Prion disease, which manifests itself as dementia, visual and cerebellar disorders and movement disorders with a fatal outcome after 4-5 months of illness in the classic variant of Creutzfeldt-Jakob disease and after (3-14 months in the new variant of Creutzfeldt-Jakob disease. The incubation period can reach 20 years. Various routes of infection and causes of the disease are possible:
- when consuming insufficiently heat-treated animal products, such as meat and brains from cows with bovine spongiform encephalopathy;
- during tissue transplantation, such as cornea transplantation, blood transfusion, use of hormones and other biologically active substances of animal origin, use of catgut, contaminated or insufficiently sterilized surgical instruments, prosectoral manipulations;
- in case of hyperproduction of PrR and other conditions that stimulate the process of converting PrR' into PrR".
The disease can also develop as a result of a mutation or insertion in the prion gene region. Familial nature of the disease is common due to genetic predisposition to Creutzfeldt-Jakob disease. In the new variant of Creutzfeldt-Jakob disease, disorders develop at a younger age (average age 28 years), in contrast to the classic variant (average age 65 years). In the new variant of Creutzfeldt-Jakob disease, abnormal prion protein accumulates not only in the central nervous system, but also in lymphoreticular tissues, including the tonsils.
Gerstmann-Sträussler-Scheinker syndrome
Hereditary prion disease, accompanied by dementia, hypotonia, swallowing disorder (dysphagia), dysarthria. Often has a familial nature. The incubation period is from 5 to 30 years. The disease occurs at 50-60 years, its duration ranges from 5 to 13 years.
Hereditary fatal insomnia
An autoimmune disease with progressive insomnia, sympathetic hyperreactivity (hypertension, hyperthermia, hyperhidrosis, tachycardia), tremor, ataxia, multiclone, hallucinations. Sleep is severely disrupted. Death occurs with progression of cardiovascular failure.
Scrape
Scrapie (from the English scrape - to scrape) is a prion disease of sheep and goats (scabies), which occurs with damage to the central nervous system, progressive movement disorders, severe skin itching (scabies) and ends in the death of the animal.
Bovine spongiform encephalopathy
A disease of cattle characterized by damage to the central nervous system, impaired coordination of movements and inevitable death of the animal. The epidemic of the disease first broke out in Great Britain. It was associated with feeding animals with meat and bone meal containing pathological prions. The incubation period ranges from 1.5 to 15 years. The brain, spinal cord and eyeballs of animals are most infected.
Laboratory diagnostics of prion diseases
During diagnostics, spongiform changes in the brain, astrocytosis (gliosis), and the absence of inflammatory infiltrates are noted. The brain is stained for amyloid. Protein markers of prion brain disorders are detected in the cerebrospinal fluid (using ELISA). Genetic analysis of the prion gene (PCR) is performed.
Prevention of prion diseases
Autoclaving (at 134°C for 18 min; at 121°C for 1 h), incineration, additional treatment with bleach and a one-normal NaCl solution for 1 h are recommended for the decontamination of instruments and environmental objects. For non-specific prophylaxis, restrictions have been introduced on the use of medicinal products of animal origin and the production of pituitary hormones of animal origin is prohibited. Transplantation of the dura mater is restricted. Rubber gloves are used when working with patients' dialogic fluids.