Medical expert of the article

New publications
Pituitary nannism (dwarfism)
Last reviewed: 12.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
The term "pituitary dwarfism" (from the Greek nanos - dwarf; syn.: dwarfism, nanosomia, microsomia) in the absolute sense means a disease, the main manifestation of which is a sharp retardation in growth, associated with a violation of the secretion of growth hormone by the anterior pituitary gland.
In a broader sense, dwarfism is a disorder of growth and physical development, the occurrence of which can be caused not only by an absolute or relative deficiency of somatotropic hormone due to pathology of the pituitary gland itself, but also by a disorder of the hypothalamic (cerebral) regulation of its functions, defects in the biosynthesis of somatotropic hormone, and disorders of tissue sensitivity to this hormone.
 [
[ Causes dwarfism
Most forms of pituitary dwarfism are genetic diseases. The most common is panhypopituitary dwarfism, which is inherited mainly in a recessive manner. It is assumed that there are 2 types of transmission of this form of pathology - autosomal and through the X chromosome. In this form of dwarfism, along with the defect in the secretion of somatotropic hormone, the secretion of gonadotropins and thyroid-stimulating hormone is most often disrupted. The secretion of ACTH is disrupted less often and to a lesser extent. Functional studies with releasing hormones, including synthetic somatotropin-releasing hormone (consisting of 29, 40 and 44 amino acid residues), similar to pancreatic polypeptide, have shown that most of these patients have pathology at the hypothalamus level, and the insufficiency of the anterior pituitary gland is secondary. Primary pathology of the pituitary gland itself is less common.
Genetic dwarfism with isolated growth hormone deficiency, with impaired biological activity and sensitivity to it, is encountered sporadically in Russia and neighboring countries. It is more common in the American continent, in the countries of the Near and Middle East, and in Africa. Based on the results of a study of the blood content of somatotropic hormone and the sensitivity of patients to exogenous somatotropic hormone, the level of immunoreactive insulin (IRI), insulin-like growth factors (IGF) type I (somatomedin C) and type II, and the response of IGF-1 to treatment with somatotropic hormone preparations, various variants of clinically similar types of dwarfism have been identified.
Recently, the pathogenesis of Laron dwarfism, which is caused by a deficiency of IRF-1 and IRF-II, has been deciphered, as well as the pathogenesis of dwarfism in African pygmies, associated with a deficiency of the former.
In 1984, a new variant of pseudo-pituitary dwarfism with a high level of somatotropic hormone and IGF-1 was described; the genesis of dwarfism is explained by a defect in its receptors, which is proven by a sharp decrease in the binding of skin fibroblasts to IGF-1.
It should be emphasized that in modern conditions, with the presence of small families, many isolated (“idiopathic”, sporadic) cases of the disease can also be genetic.
In the analysis of 350 case histories, the etiology of dwarfism was unclear in 228 patients (65.2%). This group included patients from 57 families with repeated incidence of dwarfism (2-4 cases per family), which accounted for 28% of all patients. In 77% of families with etiologically unclear (mostly genetic) forms of dwarfism, an undeniable connection with the inheritance of the absence of the Rh factor was established. The distribution of the Rh factor in families of patients with dwarfism differs from that observed in the Rh conflict between the mother and the fetus and, as a rule, is not accompanied by hemolytic disease of the newborn (the father may be Rh negative, and in the case of heterozygosity of the parents for the Rh factor - the children, etc.). It is possible to assume a connection between the activity of genes responsible for the synthesis of somatotropic hormone (or somatotropin-releasing hormone) and genes determining the Rh factor, especially since most forms of dwarfism and the absence of the Rh factor are autosomal recessive traits. This does not explain the relative rarity of dwarfism compared to the frequency of Rh-negative individuals in the population. Probably, some as yet unknown additional factors are important, but the features of the distribution of the Rh factor in families of patients with familial and sporadic dwarfism are unlikely to be accidental.
A large group of patients with dwarfism (primary cerebral, cerebral-pituitary) are patients with various types of organic pathology of the central nervous system that arose in utero or in early childhood. The anatomical substrate that causes this pathology may be underdevelopment or absence of the pituitary gland, its dystopia in pathology of the formation of the sella turcica, cystic degeneration of the pituitary gland, its atrophy due to compression by a tumor (craniopharyngioma, chromophobe adenoma, meningioma, glioma). Dwarfism may be caused by traumatic injuries to the hypothalamic-pituitary region (intrauterine, birth or postnatal), which often occurs in multiple pregnancies, as well as during childbirth in breech, foot presentation or in the transverse position with rotation on the leg (this is the mechanism of childbirth in more than 1/3 of patients with dwarfism). Infectious and toxic damages are important (intrauterine viral infections, tuberculosis, syphilis, malaria, toxoplasmosis; diseases at an early age, neonatal sepsis, meningo- and arachnoencephalitis, etc.). These processes can damage the pituitary gland itself, the hypothalamic centers that regulate its function, and disrupt normal functional connections in the central nervous system.
Intrauterine fetal lesions can lead to the birth of patients with “dwarfism from birth” with normal secretion of growth hormone (cerebral primordial dwarfism, microcephaly, Silver-Russell dwarfism with body hemi-asymmetry and high levels of gonadotropins, etc.).
Additional factors that aggravate the violation of physical development in dwarfism may be inadequate nutrition, unbalanced in terms of essential ingredients (protein deficiency) and microelements (zinc deficiency), and unfavorable environmental conditions, as well as various chronic diseases, such as glomerulonephritis, in which azotemia affects the activity of liver receptors or directly affects the metabolism of liver cells, causing a decrease in the synthesis of somatomedin, or cirrhosis of the liver, when the formation of somatomedin is impaired.
Pathogenesis
In most patients with pituitary dwarfism, changes are not limited to pathology of the secretion of somatotropic hormone and sensitivity to it, but extend to other tropic hormones of the pituitary gland, which causes various combinations of endocrine and metabolic disorders.
In isolated deficiency of somatotropic hormone, morphological changes in the pituitary gland have been poorly studied. In the cases studied, pathological disorders were rarely found (craniopharyngioma or hyperostosis of the cranial bones). In this type of dwarfism, congenital underdevelopment of peptidergic cells or a defect in the neurotransmitter system in the hypothalamus may be observed. In such cases, dwarfism may be combined with dysplasia or hypoplasia of the optic nerves. Intrasellar cysts, pituitary and hypothalamic tumors lead to STH deficiency, causing compression of the pituitary tissue, in particular somatotrophs.
Dwarfism is characterized by thinning of the bones, mainly due to the cortical layer, delayed differentiation and ossification of the skeleton. The internal organs are hypoplastic, sometimes atrophic, and the muscles are poorly developed.
Symptoms dwarfism
A sharp lag in growth and physical development are the main manifestations of pituitary dwarfism. Patients are born with normal body weight and length and begin to lag in growth from the age of 2-4 years.
Before the advent of active treatment for nanism, dwarfs were considered to be those with a height of less than 120 cm for women and 130 cm for men. Currently, the height of a dwarf differs by at least 2-3 sigma deviations from the average tabular norm for a given sex, age, or population. There is also a method for graphical assessment of height based on the Gaussian distribution curve. In this case, dwarfs by height are included in a group that includes the minimum number of individuals in the corresponding population with the greatest lag from the average growth norm.
Pituitary dwarfism is characterized not only by small absolute body size, but also by small annual growth and physical development dynamics. The body build is proportional, but the proportions of the body of patients are typical of childhood. The skin is pale, often with a yellowish tint, dry, which is due to absolute or relative thyroid insufficiency, sometimes cyanosis is observed - "marbling" of the skin. In untreated patients, old-looking and wrinkled skin (geroderma) appears early. This is due to the insufficiency of the anabolic action of STH and the slow change of cellular generations.
Hair on the head may be normal or dry, thin, brittle; long eyelashes are typical. Secondary hair growth is often absent. The size of the sella turcica in most patients with dwarfism (70-75%) is not changed, but the sella often retains the childish shape of a "standing oval", has a wide "juvenile" back, the sinus of the sphenoid bone lags behind in terms of pneumatization. However, there are patients with an enlarged sella turcica, which is a sign of a tumor; with areas of calcification on its background or in the area of the entrance (with craniopharyngioma, residual effects of neuroinfection) or its decrease (signs of underdevelopment, small size of the pituitary gland). Symptoms of intracranial hypertension are observed: thinning of the cranial vault bones, increased vascular pattern, presence of finger impressions, etc. The most important sign of pituitary dwarfism is a delay in the timing of differentiation and ossification of the skeleton. Features of the dental system are also closely related to skeletal differentiation: late replacement of milk teeth is noted. The greatest delay in the development of the skeletal system is observed in patients with dwarfism with sexual insufficiency and hypothyroidism.
The genitals of most patients are severely underdeveloped, although malformations are rare. We observed cryptorchidism in 5.8% of male patients. Sexual insufficiency is accompanied by underdevelopment of secondary sexual characteristics and decreased sexual desire, absence of menstruation. Normal spontaneous sexual development is observed only in patients with isolated growth hormone deficiency and in some patients with cerebral dwarfism.
Thyroid insufficiency is a fairly common symptom of dwarfism. It should be noted that the external manifestations of hypothyroidism do not always reflect the true functional state of the thyroid gland. This is due to relative hypothyroidism due to a violation of the transition of thyroxine (T 4 ) to triiodothyronine (T 3 ) and the formation of inactive (reversible) T 3, which is characteristic of somatotropic insufficiency.
Adrenocorticotropic function in pituitary dwarfism decreases less frequently and to a lesser extent than sex and thyroid-stimulating functions, and in most patients does not require special correction.
In most cases, the intellect is not impaired. Emotional changes in the form of mental infantilism are encountered; in older patients without intellectual impairment, reactive neuroses are sometimes observed.
In organic cerebral pathology, especially of a tumor nature, dwarfism can occur with symptoms of diabetes insipidus, bitemporal hemianopsia and intellectual disabilities.
The study of the development of bioelectrical activity of the brain in patients without organic symptoms of the central nervous system showed that their EEG is characterized by features of immaturity, long-term preservation of high "childish" EEG voltage; unevenness of the alpha rhythm in amplitude and frequency; a sharp increase in the content of slow θ- and δ-rhythms, especially in the frontal and central leads; a clear reaction to hyperventilation; a shift in the range of EEG rhythms following the rhythms of light stimulation towards low frequencies (evidence of a decrease in the functional mobility of the nervous structures of the brain). It was revealed that in older patients, the immature nature of the electrical activity of the brain is due to sexual underdevelopment, and in patients of all age groups - hypothyroidism.
Carbohydrate metabolism in patients with dwarfism is characterized by a tendency to decrease fasting blood glucose levels, an increase during physical exertion, insufficiency of endogenous insulin, increased sensitivity to exogenous insulin with frequent development of hypoglycemic conditions. The latter is explained mainly by insufficient levels of counter-insular hormones in the body of patients.
Internal organs show splanchnomycria, i.e., a decrease in their size. No functional changes in internal organs specific to dwarfism have been described. Arterial hypotension with decreased systolic and diastolic pressure and decreased pulse amplitude is often observed. Heart sounds are muffled, functional murmurs of various topics are heard due to trophic changes in the myocardium and autonomic disorders. ECG is characterized by low voltage (especially in the presence of hypothyroidism), sinus bradycardia or bradyarrhythmia; PCG shows a decrease in the amplitude of tones, additional tones, and functional murmurs. Oxygemometry data indicate hypoxemia (initial and during physical exertion), and oxygen debt. Elderly patients sometimes develop hypertension.
Diagnostics dwarfism
The diagnosis and differential diagnosis of dwarfism is based on anamnesis data and a comprehensive clinical, radiological, laboratory and hormonal examination. In addition to absolute body size, to assess the growth of patients, growth deficit is determined - the difference between the patient's height and his average norm for the corresponding sex and age; growth age - compliance of the patient's height with certain standards; the indicator of normalized deviation
I = M - Mcp / δ, where M is the patient's height, Mcp is the average normal height for a given gender and age, δ is the square deviation from Mcp; I less than 3 is typical of nanism, I more than 3 is typical of gigantism. This indicator can be used to assess the dynamics of development.
X-ray examination of patients with dwarfism reveals signs of intracranial hypertension, residual effects of neuroinfection, calcifications, and craniosynostosis. The study of the size, shape, and structure of the sella turcica is considered an indirect indicator characterizing the size of the pituitary gland. One of the most important manifestations of pathological growth retardation is a violation of skeletal differentiation. To assess the degree of skeletal maturity, bone (radiographic) age is determined, which corresponds to bone tissue differentiation; ossification deficiency is the degree of ossification lag from the norm (in years), ossification coefficient is the quotient of dividing bone age by chronological and other parameters.
Modern diagnostics of dwarfism is impossible without studying the secretion of somatotropic hormone, its basal level, circadian rhythm, and release under stimulation. Most patients with pituitary dwarfism are characterized by a reduced content of somatotropic hormone in the blood serum. When determined by the radioimmunological method, it is (according to different authors) from (0.87±0.09) to (1.50±0.64) ng/ml, with an average norm of (3.81±0.29) ng/ml. A study of daily (circadian) rhythms of somatotropic hormone secretion showed that its level in healthy people is maximum during the first 2 hours of sleep and at 4-6 am. In dwarfism, the content of somatotropic hormone is reduced during these hours as well.
To study the reserves of the somatotropic function, various stimulants are used, examining the content of somatotropic hormone before and after their introduction. Blood for the study is taken every 30 minutes for 2-3 hours. The release of somatotropic hormone after stimulation is considered normal at least up to 7-10 ng/ml, sometimes it reaches 20-40 ng/ml. If there is no reaction in one of the samples, repeat tests are carried out with other stimulants. Insufficiency of somatotropic hormone is considered proven in the absence of release of somatotropic hormone in 2-3 different samples.
The most commonly used stimulating tests are: with intravenous administration of 0.1 U (0.75-1.5 U) of insulin per 1 kg of the patient's body weight and the achievement of hypoglycemia (a decrease in the blood glucose level by 50% compared to the initial level), the serum somatotropic hormone is determined according to the above scheme. If severe hypoglycemia develops, the test is interrupted, and the patient is given glucose intravenously. This is the most common, classical diagnostic method.
TRH in a dose of 200-500 mcg intravenously. Effectively identifies hormone reserves, does not cause complications. In combination with the insulin test, it allows one to judge the level of damage to the hypothalamic-pituitary system. A positive reaction to TRH in the absence of one to insulin hypoglycemia indicates the intactness of the pituitary gland and damage at the hypothalamic level, negative reactions to TRH and hypoglycemia indicate damage to the pituitary gland itself.
TRH, LH-RH at a dose of 300 mcg intravenously is similar to the previous one.
Human SGH is a synthetic analogue of a biologically active compound isolated from a pancreatic tumor. Currently, there are 3 types of synthetic SGH: with 29, 40 and 44 amino acid residues. It is used intravenously in doses from 1 to 3 μg / kg of the patient's body weight. The release of STH is observed 15-20 minutes after administration, the test is more effective than others in revealing reserves of endogenous growth hormone. A positive SGH reaction indicates a hypothalamic level of damage to the somatotropic function and an intact pituitary gland; with amino acids (L-arginine monochloride, ornithine, tryptophan, glycine, leucine) intravenously at a dose of 0.25-0.5 g per 1 kg of the patient's body weight. Effective for studying SGH reserves. May cause allergic reactions.
L-dopa orally at a dose of 250-500 mcg. Effective, well tolerated by patients.
Tests with glucagon, bromine ergocryptine (parlodel), lysine vasopressin, clonidine, and dosed bicycle ergometric load are also used.
The study of the state of the somatotropic function is necessary not only for the diagnosis of dwarfism, but also for the justified choice of a method of therapy, since treatment with somatotropin is rational only in the case of insufficiency of endogenous growth hormone.
For the diagnosis of the form of dwarfism, it is very important to study the content of insulin-like growth factors, or somatomedins (especially IGF-1, or somatomedin C) - mediators of the action of somatotropic hormone at the tissue level. It is known that the content of somatomedin C in dwarfism is reduced, and in acromegaly - increased compared to the norm. The form of dwarfism described by Laron is a type of disease with normal production of STH, but with a violation of the formation of IGF-1 and IGF-II. Treatment of such patients with somatotropin is futile.
Indirect indicators of the somatotropic function of the pituitary gland are the activity of alkaline phosphatase and the content of inorganic phosphorus in the serum. In hyposomatotropic conditions, these indicators are reduced. In the panhypopituitary form of dwarfism, the secretion of gonadotropins, often TSH, is reduced, which is accompanied by a corresponding decrease in the functions of the sex glands (deficiency of androgens or estrogens), the thyroid gland (a decrease in the levels of T3 , T4 , protein-bound iodine - PBI, accumulation of 131 I by the thyroid gland), and the adrenal glands (a decrease in the amount of cortisol and 17-OCS in the plasma, excretion of 17-KC and 17-OCS in the urine, lymphocytosis).
All types of pituitary (hypothalamic-pituitary) genetic dwarfism are characterized by repeated disease of children in a family with inheritance by autosomal recessive (more often) or autosomal dominant type, growth retardation and physical development from 2-4 years with a lag of at least 2-3 o from the average growth norms for a given sex, age, population, with low spontaneous annual growth dynamics, delayed ossification. With a low level of somatotropic hormone (in 2-3 stimulating tests below 7 ng / ml), therapy with somatotropic hormone is highly effective (gives an increase in height of at least 7 cm per year). With a normal or high level of somatotropic hormone (with its biological inactivity), sensitivity to the hormone can be preserved. No changes in intelligence are observed
In genetic dwarfism with tissue insensitivity to somatotropic hormone, the clinical picture is similar to isolated growth hormone deficiency, but somatotropin therapy is ineffective. In this group, the following main forms can be distinguished according to the IRF level: with normal content (IRF receptor defect) and reduced - Laron type dwarfism (IRF-1 and IRF-II deficiency) and the type found in African pygmies (IRF-1 deficiency).
Cerebral dwarfism is characterized by isolated diseases in a family associated with intrauterine or postnatal damage to the central nervous system, with the presence of obvious organic changes in the central nervous system, often combined with pathology of the visual organ, the presence of diabetes insipidus, the preservation of gonadotropic functions, and changes in intelligence.
Some types of gonadal dysgenesis and ageneses are accompanied by marked short stature, in particular Shereshevsky-Turner syndrome and the "Turneroid" (mosaic) form of testicular dysgenesis syndrome. Cytogenetic studies (sex chromatin, karyotype) help in differential diagnosis, revealing chromosomal defects, as well as characteristic defects of somatic and sexual development, normal or elevated levels of endogenous somatotropic hormone, and insensitivity to treatment with somatotropin.
Among endocrine disorders occurring with short stature, primary hypothyroidism should be singled out, caused by congenital hypoplasia or aplasia of the thyroid gland, its dystopia, enzymatic defects in the biosynthesis of thyroid hormones, early autoimmune damage to the thyroid gland. In all these conditions, signs of hypothyroidism with a high level of TSH, a decrease in T4 and T3 in the blood serum dominate . In myxedema of autoimmune genesis, antibodies to thyroglobulin, microsomal and nuclear fractions of thyroid tissue are detected in the blood, the level of somatotropic hormone is normal or reduced. The clinical effect can be achieved by compensating only hypothyroidism.
Short stature is accompanied by premature sexual development and adrenogenital syndrome due to early closure of growth zones; Itsenko-Cushing's disease, which occurs in childhood due to the inhibitory effect of glucocorticoids on the secretion of somatotropic hormone and their catabolic effect; Mauriac syndrome - short stature and infantilism of patients with severe insulin-dependent diabetes mellitus.
Pituitary dwarfism should be differentiated from somatogenic delay in physical development caused by chronic metabolic disorders (in diseases of the liver, kidneys, gastrointestinal tract), chronic hypoxia (in diseases of the cardiovascular and respiratory systems, in anemia); with systemic diseases of the musculoskeletal system (chondrodystrophy, imperfect osteogenesis, exostosis disease), etc.
Functional (constitutional) growth retardation is sometimes observed with late onset of puberty in apparently healthy adolescents; we have found that it is primarily associated with transient insufficiency of gonadotropic activity. Secretion of somatotropic hormone is usually not impaired or is slightly reduced. Stimulation of gonadotropins can accelerate both sexual development and growth.
Short stature of a family nature should be considered as a variant of physiological development.
What do need to examine?
How to examine?
What tests are needed?
Who to contact?
Treatment dwarfism
Treatment of dwarfism is a long process. This forces the doctor to distribute the means of influencing growth over time in order to obtain the greatest clinical effect while observing 2 basic principles:
- maximum approximation of treatment-induced development to physiological conditions;
- sparing the epiphyseal growth zones.
Many years of experience in treating dwarfism allow us to consider the following scheme of staged therapy to be appropriate. The diagnosis of dwarfism in adult patients usually does not raise doubts. In small children, if the clinical picture is unclear, a diagnostic period is necessary: 6-12 months under observation without hormone therapy. During this time, complex general strengthening treatment is prescribed; adequate nutrition with an increase in the content of animal protein, vegetables and fruits in the diet, vitamins A and D, calcium and phosphorus preparations. The absence of sufficient changes in growth and physical development against this background and the detection of endocrine disorders during examination are the basis for starting hormone therapy.
The main type of pathogenetic therapy for pituitary dwarfism is the use of human growth hormone, since the occurrence of most cases of dwarfism is undoubtedly dependent on one or another form of its deficiency. Due to the species specificity of this hormone, only human and primate somatotropin is active for humans. A drug isolated from the pituitary gland of people who died from non-infectious and non-neoplastic diseases is widely used in the clinic. Human somatotropin is obtained by bacterial synthesis using Escherichia coli by genetic engineering. Human somatotropin is also synthesized chemically, but it is extremely expensive and is practically not used in the clinic. Patients with proven deficiency of endogenous growth hormone, with skeletal differentiation not exceeding the level typical of 13-14 years old, are selected for somatotropin treatment. There are no age restrictions for treatment.
The minimum effective doses that can be used in the first period of treatment are 0.03-0.06 mg/kg of body weight. The most effective doses are 2-4 mg 3 times a week. Increasing the single dose to 10 mg was not accompanied by an adequate increase in the growth effect, but caused rapid formation of antibodies to somatotropin.
In our country, work on studying human growth hormone has been conducted since 1960. Two treatment regimens have been tested: continuous and intermittent with courses of 2-3 months and the same intervals between them. The average increase in height of patients during the 1st year of treatment was 9.52±0.39 cm, the increase in body weight was 4.4±0.14 kg. With long-term continuous treatment, the average increase in height was 0.82 cm/month, body weight - 0.38 kg/month; with intermittent - 0.75 cm/month and 0.4 kg/month, respectively. Continuous treatment provided a more rapid increase in height with a sharp decrease in the effect after 1-1.5 years, with intermittent treatment, the effectiveness was maintained for 3-4 years, which allows us to consider the course treatment regimen more appropriate. Determination of the level of IGF-I (somatomedin C) can serve as a reliable indicator of the patient's sensitivity to treatment with somatotropin drugs. An increase in the IGF-I content after the introduction of somatotropic hormone allows one to predict a positive effect of therapy. An important advantage of somatotropin treatment is the absence of acceleration of skeletal ossification against its background.
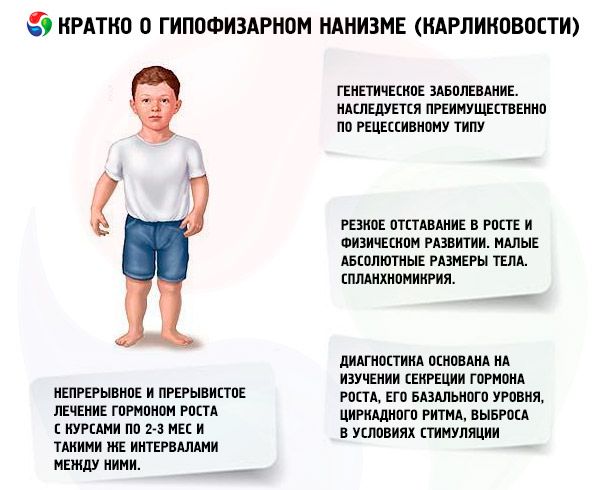
The most important means of treating dwarfism is the use of anabolic steroids, which stimulate growth by enhancing protein synthesis and increasing the level of endogenous somatotropic hormone. Treatment is carried out for several years, with a gradual replacement of some drugs with others, from less active to more active compounds. A change in anabolic drugs is indicated when the growth effect decreases after 2-3 years, which leads to an additional increase in growth. Treatment is carried out in courses (the rest period should be half the treatment period). In case of addiction, longer breaks are also indicated (up to 4-6 months). Only one of the anabolic steroids is prescribed at a time. Combining 2 or more drugs is inappropriate, since this does not enhance their metabolic and growth effects. The latter depends primarily on the age of the patients and the degree of differentiation of the skeletal bones at the beginning of treatment. The best effect is observed in patients under 16-18 years of age with skeletal ossification not exceeding the level characteristic of a 14-year-old. It is advisable to begin treatment immediately after diagnosis, usually from the age of 5-7 years. Before treatment, it is necessary to avoid prescribing gonadotropins and sex hormones, which, while stimulating growth, simultaneously accelerate skeletal differentiation. The principle of dosing anabolic steroids is from the minimum effective doses to gradually increasing ones. The recommended doses of the most common drugs: nerobol (methandrostenol, dianabol) - 0.1-0.15 mg per 1 kg of body weight per day orally; nerobolil (durabolin) - 1 mg per 1 kg of body weight per month intramuscularly, the monthly dose is administered in 2-3 doses, respectively, after 15 or 10 days; retabolil (deca-durabolin) - 1 mg per 1 kg of body weight once a month intramuscularly. Exceeding the specified doses can lead to androgenization. In physiological doses, these compounds do not significantly affect the condition of the genitals and differentiation of skeletal bones, which allows them to be used for a long time in patients of both sexes. Girls should be under the supervision of a gynecologist, since in case of an overdose or increased individual sensitivity, some patients may develop signs of virilization, which quickly regress when treatment is discontinued. Oral drugs methylated to ethylated in the 17th position can sometimes cause a cholestatic effect, therefore, in liver diseases, preference should be given to parenteral anabolic compounds, or oral drugs should be combined with choleretic agents. Very rarely, treatment with anabolic steroids can cause allergic reactions (itching, rashes). In the absence of complications, anabolic steroids are used as long as the growth effect is observed (up to 16-18 years, and sometimes longer). Treatment is carried out against the background of general strengthening therapy.
If patients have signs of hypothyroidism, thyroid medications (thyroxine, thyroidin, thyrotom) are prescribed simultaneously in individually selected doses.
In the treatment of boys, the next step is the administration of human chorionic gonadotropin. This drug is used no earlier than 15-16 years, and often at an even later age to stimulate Leydig cells, which accelerates both sexual development and growth (due to the anabolic activity of their own androgens). Doses of 1000 to 1500 IU are used 1-2 times a week intramuscularly in 2-month courses no more than 2-3 times a year. If the effect is incomplete, treatment with human chorionic gonadotropin in boys aged 16 years and older is alternated with the administration of small doses of androgens (methyltestosterone at a dose of 5-10 mg/day sublingually).
Girls over 16 years of age can begin treatment with small doses of estrogens, simulating a normal sexual cycle. Treatment is carried out for 3 weeks of each month, followed by a break. In the 2nd phase of the cycle, from the 3rd week, chorionic gonadotropin can be prescribed at a dose of 1000-1500 IU 3-5 times a week or drugs with a gestagenic effect (pregnin, progesterone).
The final stage of treatment (after the closure of growth zones) is the constant administration of therapeutic doses of sex hormones corresponding to the patient's gender, in order to fully develop the genitals, secondary sexual characteristics, ensure libido and sexual potency. Combined estrogen-progestogen preparations (non-ovlon, bisecurin, infekundin, rigevidon) are convenient for treating female patients, and prolonged-release androgen preparations (testenate, sustanon-250, omnadren-250) are convenient for treating male patients.
General strengthening treatment is carried out (regime, protein-vegetable diet, vitamin therapy, biostimulants). The use of zinc preparations is indicated, in the mechanism of action of which the main role is played by increasing the activity of IGF-1 (insulin-like growth factor I).
In the presence of organic pathology from the central nervous system, anti-inflammatory, resorptive, and dehydration therapy are administered. Targeted systematic therapy gives an encouraging effect. As a result of long-term staged treatment, 148 (80.4%) of 175 patients with dwarfism of both sexes managed to achieve a height of over 130 cm, 92 (52.5%) - over 140 cm, and 32 (18.3%) - 150-160 cm or more. At the same time, 37 patients (21.2%) increased their height by 30 cm, 107 (61.1%) by 31-50 cm, and 31 (17.7%) by 51-60 cm or more.
Forecast
The prognosis depends on the form of dwarfism. In genetic types of dwarfism, the prognosis for life is favorable. In the presence of a pituitary tumor and organic damage to the central nervous system, it is determined by the dynamics of the development of the main pathological process. Modern methods of therapy have significantly increased the physical capabilities and working capacity of patients, and extended their life expectancy. During the period of active treatment, patients need to be examined by a doctor every 2-3 months, with maintenance therapy - every 6-12 months.
Employment of patients that corresponds to their intellectual and physical capabilities is of paramount importance for their social adaptation.
It is advisable to choose professions that are not associated with heavy physical exertion, but allow you to demonstrate intellectual abilities, the ability to do precise work, and languages.