Medical expert of the article

New publications
Charcot-Marie-Tooth disease.
Last reviewed: 12.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
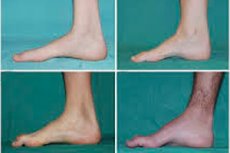
Peroneal muscular atrophy, Charcot-Marie-Tooth syndrome or disease is a group of chronic hereditary diseases with damage to the peripheral nerves.
According to ICD-10, in the section on diseases of the nervous system, the code for this disease is G60.0 (hereditary motor and sensory neuropathy). It is also included in the list of orphan diseases.
Epidemiology
According to clinical statistics, the prevalence of all types of Charcot-Marie-Tooth disease per 100 thousand of the population is 19 cases (according to other sources, one case per 2.5-10 thousand of the population).
CMT type 1 accounts for about two-thirds of cases (one case per 5,000 to 7,000 population), and nearly 70% of them are associated with duplication of the PMP22 gene. More than 1.2 million people worldwide suffer from this type of disease.
The incidence of CMT type 4 is estimated at 1-5 cases per 10,000 children. [ 1 ]
Causes Charcot-Marie-Tooth disease
According to the classification of polyneuropathic syndromes, peroneal (fibular) muscular atrophy, Charcot-Marie-Tooth neural amyotrophy or Charcot-Marie-Tooth disease (abbreviated as CMT) refers to genetically determined motor-sensory polyneuropathies. [ 2 ]
That is, the causes of its occurrence are genetic mutations. And depending on the nature of genetic deviations, the main types or kinds of this syndrome are distinguished: demyelinating and axonal. The first group includes Charcot-Marie-Tooth disease type 1 (CMT1), which occurs due to duplication of the PMP22 gene on chromosome 17, which codes for transmembrane peripheral myelin protein 22. As a result, segmental demyelination of the axon sheath (nerve cell processes) and a decrease in the speed of nerve signal conduction occur. In addition, mutations may be in some other genes.
The axonal form is Charcot-Marie-Tooth disease type 2 (CMT2), which affects the axons themselves and is associated with pathological changes in the MFN2 gene at locus 1p36.22, encoding the membrane protein mitofusin-2, which is necessary for mitochondria fusion and the formation of functional mitochondrial networks inside peripheral nerve cells. There are more than a dozen subtypes of CMT2 (with mutations in specific genes).
It should be noted that currently more than a hundred genes have been identified whose damage, transmitted by inheritance, causes various subtypes of Charcot-Marie-Tooth disease. For example, mutations in the RAB7 gene lead to CMT type 2B; alteration of the SH3TC2 gene (encoding one of the membrane proteins of Schwann cells) causes CMT type 4C, which manifests itself in childhood and is characterized by demyelination of motor and sensory neurons (there are a dozen and a half forms of type 4 of this disease).
A rare type 3 CMT (called Dejerine-Sottas syndrome) begins to develop in early childhood and is caused by mutations in the PMP22, MPZ, EGR2, and other genes.
When CMT type 5 occurs at the age of 5-12 years, not only motor neuropathy (in the form of spastic paraparesis of the lower extremities) is observed, but also damage to the optic and auditory nerves.
Muscle weakness and optic atrophy (with vision loss) as well as balance problems are typical of CMT type 6. And in type 7 Charcot-Marie-Tooth disease there is not only motor-sensory neuropathy, but also a retinal disease in the form of retinitis pigmentosa.
More common among males, X-linked CMT or Charcot-Marie-Tooth disease with tetraparesis of the limbs (weakness of movement of both arms and legs) is a demyelinating type and is thought to result from a mutation in the GJB1 gene on the long arm of the X chromosome, which encodes connexin 32, a transmembrane protein of Schwann cells and oligodendrocytes that regulates the transmission of nerve signals. [ 3 ]
Risk factors
The main risk factor for CMT is a family history of the disease, that is, in close relatives.
According to geneticists, if both parents are carriers of the autosomal recessive gene for Charcot-Marie-Tooth disease, the risk of having a child who will develop this disease is 25%. And the risk that the child will be a carrier of this gene (but will not have any symptoms) is estimated at 50%.
In the case of X-linked inheritance (when the mutated gene is on the woman's X chromosome), there is a 50% risk that the mother will pass the gene on to her son, who will develop CMT. The disease may not occur when a female child is born, but the daughter's sons (grandchildren) may inherit the defective gene, and the disease will develop.
Pathogenesis
In any type of Charcot-Marie-Tooth disease, its pathogenesis is caused by a hereditary anomaly of the peripheral nerves: motor (movement) and sensory (sensitive).
If the CMT type is demyelinating, then the destruction or defect of the myelin sheath that protects the axons of the peripheral nerves leads to a slowdown in the transmission of nerve impulses in the peripheral nervous system - between the brain, muscles and sensory organs.
In the axonal type of the disease, the axons themselves are affected, which negatively affects the strength of nerve signals, which is insufficient for full stimulation of muscles and sensory organs.
Also read:
How is Charcot-Marie-Tooth syndrome transmitted? The defective genes can be inherited in an autosomal dominant or autosomal recessive manner.
The most common type, autosomal dominant inheritance, occurs when there is one copy of the mutated gene (carried by one of the parents). And the probability of passing on CMT to each child born is estimated at 50%. [ 4 ]
With autosomal recessive inheritance, two copies of the defective gene (one from each parent who does not show any signs of the disease) are needed for the disease to develop.
In 40-50% of cases, autosomal dominant hereditary demyelination occurs, i.e. CMT type 1; in 12-26% of cases, axonal CMT, i.e. type 2. And in 10-15% of cases, X-linked inheritance is observed. [ 5 ]
Symptoms Charcot-Marie-Tooth disease
Typically, the first signs of this disease begin to appear in childhood and adolescence and gradually develop throughout life, although the syndrome may make itself known later. The combination of symptoms is variable, and the rate of progression of the disease, as well as its severity, is impossible to predict.
Typical symptoms of the initial stage include increased general fatigue; decreased tone (weakness) of the muscles of the feet, ankles and shins; lack of reflexes. This makes foot movement difficult and leads to dysbasia (gait disturbance) in the form of a higher rise of the legs, often with frequent trips and falls. Signs of Charcot-Marie-Tooth disease in a small child may include pronounced clumsiness and age-uncharacteristic walking difficulties associated with bilateral foot drop. Foot deformities are also characteristic: high arch (hollow foot) or severe flat feet, curved (hammer-shaped) toes.
In the case of walking on the toes against the background of muscle hypotonia, a neurologist may suspect that the child has type 4 CMT, in which children may be unable to walk by adolescence.
As the disease progresses, muscle atrophy and weakness spread to the upper extremities, making it difficult to perform fine motor skills and perform normal hand tasks. Decreased tactile sensations and the ability to feel heat and cold, as well as numbness in the feet and hands, indicate damage to the axons of the sensory nerves.
In Charcot-Marie-Tooth disease types 3 and 6 that manifests in childhood, sensory ataxia (impaired coordination of movements and balance), muscle twitching and tremor, damage to the facial nerve, optic nerve atrophy with nystagmus, and hearing loss are observed.
In later stages, there may be uncontrollable shaking (tremor) and frequent muscle cramps; problems with movement can lead to the development of pain: muscle, joint, neuropathic.
Complications and consequences
Charcot-Marie-Tooth disease can have complications and consequences such as:
- more frequent sprains and fractures;
- contractures associated with shortening of periarticular muscles and tendons;
- scoliosis (curvature of the spine);
- breathing problems – when the nerve fibers that innervate the muscles of the diaphragm are damaged:
- loss of the ability to move independently.
Diagnostics Charcot-Marie-Tooth disease
Diagnosis includes clinical examination, anamnesis (including family history), neurological and systemic examination.
Tests are performed to check range of motion, sensitivity and tendon reflexes. Nerve conduction can be assessed using instrumental diagnostics – electromyography or electroneuromyography. Ultrasound or MRI may also be required. [ 6 ]
Genetic or DNA testing to detect the most common genetic mutations that cause CMT in a blood sample is limited because DNA tests are not currently available for all types of the disease. For more information, see Genetic testing
In some cases, a biopsy of the peripheral nerve (usually the sural nerve) is performed.
Differential diagnosis
Differential diagnosis includes other peripheral neuropathies, Duchenne muscular dystrophy, myelopathic and myasthenic syndromes, diabetic neuropathy, myelopathies in multiple and amyotrophic lateral sclerosis, Guillain-Barré syndrome, trauma to the peroneal nerve and its atrophy (including when pinched between the lumbar discs of the spine), damage to the cerebellum or thalamus, as well as side effects of chemotherapy (during treatment with cytostatics such as Vincristine or Paclitaxel). [ 7 ]
Who to contact?
Treatment Charcot-Marie-Tooth disease
Today, treatment for this hereditary disease includes exercise therapy (aimed at strengthening and stretching muscles); occupational therapy (which helps patients with muscle weakness in the hands); and the use of orthopedic devices to facilitate walking. If necessary, painkillers or anticonvulsants are prescribed. [ 8 ]
In cases of severe flat feet, osteotomy can be performed, and in case of heel deformation, surgical correction is indicated – arthrodesis. [ 9 ]
Research is underway into both the genetic component of the disease and its treatment methods. The use of stem cells, certain hormones, lecithin or ascorbic acid have not yet yielded positive results.
But thanks to recent research, something new may indeed appear in the treatment of Charcot-Marie-Tooth disease in the near future. Thus, since 2014, the French company Pharnext has been developing, and since mid-2019, clinical trials have been underway for the drug PXT3003 for the treatment of CMT type 1 in adults, which suppresses increased expression of the PMP22 gene, improves myelination of peripheral nerves and alleviates neuromuscular symptoms.
Specialists from the medical company Sarepta Therapeutics (USA) are working on creating a gene therapy for type 1 Charcot-Marie-Tooth disease. This therapy will use a harmless adeno-associated virus (AAV) of the Dependovirus genus with a linear single-stranded DNA genome, which will transfer the NTF3 gene into the body, encoding the neurotrophin-3 (NT-3) protein necessary for the functioning of Schwann nerve cells.
Helixmith will begin clinical trials of the South Korean-developed gene therapy Engensis (VM202) by the end of 2020 to treat muscle symptoms in CMT type 1. [ 10 ]
Prevention
Prevention of CMT can be genetic counseling of future parents, especially if someone in the couple has a family history of the disease. However, cases of de novo point gene mutations have been identified, that is, in the absence of the disease in the family history.
During pregnancy, the probability of Charcot-Marie-Tooth disease in the future child can be checked by chorionic villus biopsy (from 10 to 13 weeks of gestation), as well as an analysis of amniotic fluid (at 15-18 weeks).
Forecast
In general, the prognosis for different types of Charcot-Marie-Tooth disease depends on the clinical severity, but in all cases the disease progresses slowly. Many patients have disabilities, although this does not reduce life expectancy.