Medical expert of the article

New publications
Parietal meningioma
Last reviewed: 29.06.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
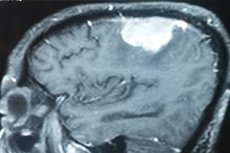
Parietal meningioma or parietal meningioma is a tumor that originates from modified meningothelial cells of the middle dura mater with attachment to the inner layer of the dura mater over the parietal lobes (lobus parietalis) of the cerebral cortex. Most tumors of this type (80-90%) are benign.
Epidemiology
Meningiomas account for 37.6% of all primary CNS tumors and 53.3% of nonmalignant intracranial tumors; multiple meningiomas occur in less than 10% of cases. They are most common in adults 40-60 years of age, and are rarely detected in children. These neoplasms are three times more common in women than in men.
Grade II meningiomas account for up to 5-7% of cases, while grade III meningiomas account for 1-2%.
Parietal meningioma is a fairly rare diagnosis.
Causes of the parietal meningiomas
Meningioma is considered the most common primary intracranial tumor, it is formed by pathologically overgrown meningothelial cells of the spider web (arachnoidea mater encephali) of the brain. [1]
Typically, meningiomas occur spontaneously, meaning the causes are unknown.
Brain tumors, including intracranial meningiomas, are thought to arise from chromosomal abnormalities and defects (mutations, aberrations, splicing, amplification or loss) of genes regulating the rate of cell division (due to protein growth factors) and the process of cell apoptosis, tumor suppressor genes, etc. The tumor suppressor genes are also thought to arise from chromosomal abnormalities and defects (mutations, aberrations, splicing, amplification or loss) of genes regulating the rate of cell division (due to protein growth factors) and the process of cell apoptosis.
For example, a genetic disorder such as loss of chromosome 22q results in a familial syndrome, neurofibromatosis type 2, which accounts not only for many cases of meningiomas but also for an increased incidence of other brain tumors.
Meningiomas are divided into three grades: benign (grade I), atypical (grade II), and anaplastic or malignant (grade III). Histologic varieties of meningiomas are also distinguished: fibrous, psammomatous, mixed, etc.
Risk factors
To date, the only proven factor that increases the risk of developing meningiomas is exposure to ionizing radiation (radiation) to the head region (especially in childhood).
They also found an association between the development of this type of tumor and obesity, which the researchers attributed to increased signaling of insulin and insulin-like growth factor (IGF-1), which inhibit cell apoptosis and stimulate tumor growth.
Some researchers have noted an increased risk of meningioma in people occupationally associated with the use of pesticides and herbicides.
Pathogenesis
The meningothelial cells of the spider web originate from germinal tissue (mesenchyme); they form dense intercellular contacts (desmoses) and create two barriers at once: between cerebrospinal fluid and nervous tissue and between the liquor and the circulation.
These cells are lined by the spider and soft cerebral membrane (pia mater encephali), as well as spider septa and ties that cross the subarachnoid space, a space filled with cerebrospinal fluid between the spider and soft cerebral membranes.
The molecular mechanism of increased proliferation of enveloped meningothelial cells and the pathogenesis of sporadic meningioma formation are poorly understood.
A benign tumor (grade I meningioma) that has a distinct rounded shape and base, the cells that form it do not grow into the surrounding brain tissue, but usually grows inside the skull and puts focal pressure on adjacent or lower brain tissue. Tumors can also grow outward, causing thickening of the skull (hyperostosis). In anaplastic meningiomas, growth may be invasive (spreading to the cerebral tissue).
Histological studies have shown that many tumors of this type have an area with the highest proliferative activity. And there is a hypothesis according to which meningiomas are formed from a certain neoplastic transformed cell clone that spreads through the brain membranes.
Symptoms of the parietal meningiomas
It is difficult to notice the first signs of meningioma of the parietal region, since headaches are a nonspecific symptom and do not occur in everyone, and the tumor itself grows slowly.
If symptoms occur, their nature and intensity depend on the size and location of the meningioma. In addition to headaches and dizziness, they may manifest as epileptiform convulsions, visual impairment (blurred vision), weakness in the extremities, sensory disturbances (numbness), loss of balance.
When a left parietal meningioma develops, patients experience: forgetfulness, unsteady gait, difficulty swallowing, right-sided motor weakness with unilateral muscle paralysis (hemiparesis), and problems with reading (alexia).
A right parietal meningioma, which may form between the right parietal lobes and the soft dura (located beneath the dura), initially presents with headaches and bilateral weakness in the extremities. Swelling near the tumor and/or compression of the tumor mass on the parietal region may cause double vision or blurred vision, tinnitus and hearing loss, loss of sense of smell, seizures, and speech and memory problems. With increased compression, some symptoms of parietal lobe lesions also develop, including parietal association cortex deficits with attention or perceptual deficits; astereognosis and problems with orientation; and contralateral apraxia - difficulty performing complex motor tasks.
A convex or convexital parietal meningioma grows on the surface of the brain, and more than 85% of cases are benign. Signs of such a tumor include headaches, nausea and vomiting, motor slowing, and frequent muscle seizures in the form of partial seizures. There may be erosion or hyperostosis (thickening of the skull) in the cranial bone in contact with the meningioma, and there is often a zone of calcification at the base of the tumor, which is defined as calcified meningioma of the parietal lobe.
Complications and consequences
Tumor enlargement and its pressure can lead to increased intracranial pressure, damage to cranial nerves (with the development of various neurological disorders), displacement and compression of parietal gyrus (which can cause psychiatric abnormalities).
Metastasis is an extremely rare complication seen in grade III meningiomas.
Diagnostics of the parietal meningiomas
Diagnosis of these tumors involves an exhaustive patient history and neurological examination.
Blood and cerebrospinal fluid tests are necessary.
The main role in tumor detection is played by instrumental diagnostics: brain CT with contrast, brain MRI, MP-spectroscopy, positron emission tomography (PET scan), CT-angiography of cerebral vessels. [2]
Differential diagnosis
Differential diagnosis includes meningothelial hyperplasia, cerebral tuberculosis, glioma, schwannoma, hemangiopericytoma, and all intracranial mesenchymal tumors.
Who to contact?
Treatment of the parietal meningiomas
For parietal meningioma, treatment is aimed at reducing compression of the brain and removing the tumor.
But if the tumor does not cause symptoms, it does not require immediate treatment: specialists monitor its "behavior" with periodic MRI scans.
For intracranial meningiomas chemotherapy is rarely used if the tumor is grade III or has recurred. In the same cases, radiation therapy with stereotactic radiosurgery and intensity modulated proton therapy is performed.
Drug treatment, that is, the use of medicine, may include: taking an antineoplastic agent in capsules such as Hydroxyurea (hydroxycarbamide); injections of an antitumor hormone sandostatin. Immunotherapy by administering alpha interferon (2b or 2a) preparations may be given.
Medications are also prescribed to relieve some symptoms: corticosteroids for swelling, anticonvulsants for seizures, etc.
When a meningioma causes symptoms or grows in size, surgical treatment - subtotal resection of the tumor - is often recommended. During surgery, samples of tumor cells (biopsy) are taken for histological examination - to confirm the type and extent of the tumor. Although complete removal can provide a cure for meningioma, this is not always possible. The location of the tumor determines how safe it will be to remove it. And if part of the tumor remains, it is treated with radiation.
Meningiomas sometimes recur after surgery or radiation, so regular (every one to two) MRIs or CT scans of the brain are an important part of treatment.
Prevention
There is no way to prevent meningioma formation.
Forecast
The most reliable prognostic factor for parietal meningioma is its histologic grade and the presence of recurrence.
While the 10-year overall survival rate for grade I meningiomas is estimated at almost 84%, for grade II tumors it is 53% (with fatal outcome in cases of grade III meningiomas). And the recurrence rate within five years after appropriate treatment in patients with benign meningiomas averages 15%, with atypical tumors - 53%, and with anaplastic - 75%.