Medical expert of the article

New publications
Angelman syndrome in children and adults
Last reviewed: 04.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.

There are a number of diseases for which expressions like "take care of yourself and you won't get sick" sound, at the very least, ridiculous. These are pathologies in which some mental and physical abnormalities are inherent in the child's body even before birth, but the parents are not to blame for this. Such diseases are caused by mutations or abnormalities in chromosome sets and are called chromosomal or genetic. Angelman syndrome, Down syndrome, Patau syndrome, Edwards syndrome, Turner syndrome, Prader-Willi syndrome - this is only part of the genetic diseases from a fairly decent list.
Happy Man Syndrome
This time we will talk about the pathology named after the English pediatrician Harry Angelman, who first raised the issue of this problem in 1965, having encountered three unusual children in his practice the day before, united by common peculiar symptoms. The doctor called these children doll children and wrote an article about them, which was initially called "Children-marionettes". The article itself and its title were written under the impression of a painting seen in one of the museums of Verona. The painting depicted a laughing boy, and it was called "The Puppet Boy". The association of the child depicted in the painting with the three children that Angelman once encountered in his practice prompted the pediatrician to combine the children into one group due to the disease they had.
There is nothing surprising in the fact that the children mentioned in the article were not noticed by other doctors. After all, at first glance it seemed that they had completely different diseases, so different was the general clinical picture of the disease in 3 different cases. Perhaps the "new" chromosomal pathology would have interested other scientists, but at that time genetics was not yet developed enough to confirm the hypothesis of the English doctor. Therefore, after a certain interest in it, the article was thrown on the back shelf for a long time.
The next mention of Angelman syndrome, which is what the article by the English pediatrician G. Angelman was now called, dates back to the early 80s of the 20th century. And only in 1987 was it possible to find the reason why a small part of children are born with such deviations that from the outside they seem to be constantly smiling and happy. In fact, this is not true at all, and the smile is just a grimace, behind which hides an unhappy human soul and the pain of the parents.
Epidemiology
According to statistics, a chromosomal mutation in a child can develop both against the background of similar mutations in parents and in the absence of such. There is no clear hereditary nature of Angelman syndrome (AS), but the probability of developing pathology in parents with chromosomal mutations is quite high.
It is also interesting that if a family already has a child with AS, there is a one percent chance of having a second child with the same disorder, even if the parents are healthy.
There are still no exact statistics on the number of patients with Angelman syndrome. Perhaps the reason is the variety of symptoms, which may occur in a certain composition or not occur at all for a long time. It is assumed that the prevalence of the disease is: 1 child per 20,000 newborns. But this figure is very approximate.
Causes Angelman syndrome
Angelman syndrome is a medical name for a chromosomal pathology, but it is far from the only one. People call this disease doll children syndrome, happy puppet syndrome, Petrushka syndrome, and laughing doll syndrome. People come up with all sorts of names (sometimes even offensive to the patients themselves and their parents), but a disease is a disease, no matter how funny it may look and no matter what the reasons are.
And the reasons for the development of Angelman syndrome, like many other genetic pathologies, in all cases are disturbances in the structure of one of the chromosomes or the chromosome set as a whole. But in our case, the whole problem lies in chromosome 15, passed on from the mother. That is, the paternal chromosome in this case has no deviations, but the female one undergoes certain mutations.
According to the type of chromosomal abnormality, Angelman syndrome is classified as a chromosomal mutation. Such mutations are considered to be:
- A deletion (absence of a section of a chromosome containing a certain set of genes; if one of the genes is missing, we are talking about a microdeletion), which is the result of two breaks and one reunion, when a section of the original chromosome is lost.
- Duplication (the presence of an extra section in a chromosome that is a copy of an existing one), which in most cases leads to death of a person, and less often to infertility.
- Inversion (reversal of one of the sections of the chromosome by 180 degrees, i.e. in the opposite direction, and then the genes in it are located in the opposite order), when the broken ends of the chromosome are connected in an order different from the original.
- Insertion (if part of the genetic material in a chromosome is out of place),
- translocation (if a certain section of a chromosome is attached to another chromosome; such a mutation can be mutual without loss of sections).
Receiving a mutated chromosome from an unsuspecting mother, the child is doomed to be born with abnormalities. The most common cause of Angelman syndrome is still considered to be a deletion of the maternal 15th chromosome, when a small section is missing. Less common mutations in the "laughing doll" syndrome are considered to be:
- translocation,
- unipaternal disomy (if the child received a pair of chromosomes from the father, the maternal chromosome is absent),
- mutation of genes in DNA, which are both the main building (genetic) material and instructions for its correct use (in particular, mutation of the ube3a gene in the maternal chromosome).
The presence of one of these mutations in parents is a risk factor for the development of Angelman syndrome in children. But not only chromosomal mutations, but also genomic ones (which are associated with a quantitative change in chromosome sets and are more common than chromosomal ones) can provoke the development of the disease in a child. Common genomic mutations include chromosome trisomy (if a person's chromosome set has more than 46 chromosomes).
For a pathology to appear in a child, it is not at all necessary for the parents to have chromosomal abnormalities. And yet, there is a certain percentage of patients whose disease is hereditary.
Pathogenesis
Let's delve a little deeper into biology, or more precisely, genetics. The genetic information of each individual human organism is contained in 23 pairs of chromosomes. One chromosome from a pair is passed on to the child from the father, the other from the mother. All pairs of chromosomes differ in shape and size and carry certain information. Thus, the 23rd pair of chromosomes (X and Y chromosomes) is responsible for the formation of the baby's sexual characteristics (XX - girl, XY - boy, while the Y chromosome can only be received by the child from the father).
Ideally, a child receives 46 chromosomes from his parents, which form his genetic characteristics, predetermining him as an individual. A larger number of chromosomes is called trisomy and is considered a deviation from the norm. For example, the presence of chromosome 47 in the chromosome set (karyotype, determining species and individual characteristics) causes the occurrence of Down syndrome.
If the chromosomes are stained with a special dye, then under the microscope you can see stripes of different shades along each of them. Inside each stripe there is a huge number of genes. All these stripes are numbered by scientists and have a fixed location. The absence of one of the stripes is considered a deviation from the norm. In Angelman syndrome, one can very often observe the absence of segments of the maternal chromosome in the interval q11-q13, located in the long arm, the number of DNA bases in which is only about 4 million.
The main component of the chromosome is considered to be an incredibly long DNA molecule containing thousands of genes and tens and hundreds of millions of nitrogenous bases. Thus, chromosome 15, responsible for the development of Angelman syndrome and several others, contains 1200 genes and about 100 million bases. Any disturbances in the structure of the DNA molecule will certainly affect the appearance and development of the future child.
The genetic information contained in genes is converted into protein or RNA. This process is called gene expression. In this way, the genetic information received from parents receives both form and content, which is embodied in their unique female or male heir.
There are a number of pathologies with a non-classical type of inheritance, including Angelman syndrome, in which genes received from parents as part of paired chromosomes bear a unique imprint of the parents and manifest themselves in different ways.
So, Angelman syndrome is a striking example of genomic imprinting, in which gene expression in the child's body is directly dependent on which parent the alleles were received from (different forms of one gene, received from the father and mother, located on identical sections of paired chromosomes). That is, only anomalies in the maternal chromosome lead to the development of the syndrome, while mutations and structural disorders of the paternal chromosome cause completely different pathologies.
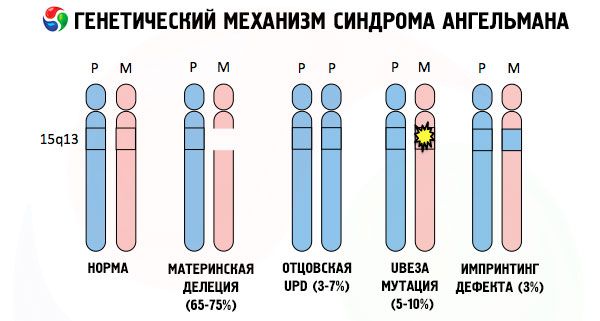
In this pathology, there is a lack of certain genes in the maternal chromosome or a loss/reduction in the activity of individual genes (in the vast majority of cases, the ube3a gene, which is involved in the metabolism of ubiquitin, a protein that regulates the degradation of other proteins). As a result, the child is diagnosed with mental developmental abnormalities and physical deformities.
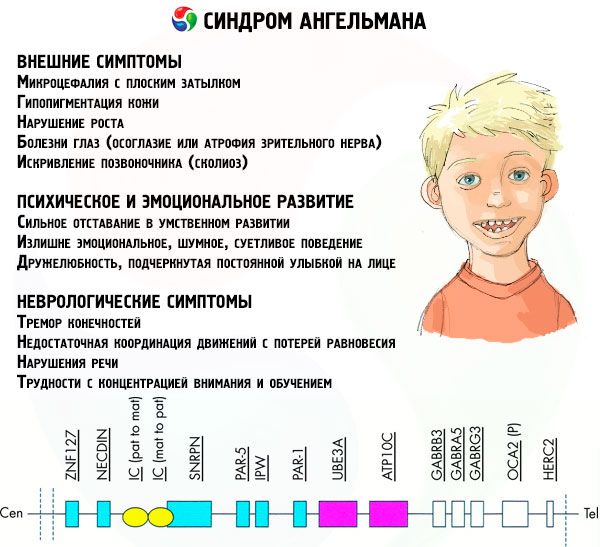
Symptoms Angelman syndrome
The symptoms of Angelman syndrome affect various aspects of a child's life and development: physical, neurological, mental. Based on this, 3 groups of symptoms can be identified that indicate the development of this pathology.
- External or physical symptoms:
- a disproportionately small head compared to the body and limbs, which are of normal size,
- too wide mouth,
- there is almost always a smile on the face (with an open mouth),
- sparse teeth,
- narrow upper lip,
- frequently protruding wide tongue,
- protruding lower jaw,
- pointed chin,
- very light skin, often hair (albinism, associated with the fact that the body does not produce the pigment melanin),
- dark spots on fair skin (hypopigmentation due to insufficient melanin production)
- physical or external symptoms: eye diseases such as strabismus or optic nerve atrophy,
- curvature of the spine (scoliosis),
- stiff legs (when walking, a person does not bend his legs at the knees due to low mobility of the joints, hence the comparison with a doll's gait).
- Symptoms related to mental and emotional development:
- severe mental retardation,
- overly emotional, noisy, fussy behavior,
- frequent clapping of hands,
- expressed friendliness, emphasized by a constant smile on the face,
- frequent laughter for no reason.
- Neurological symptoms:
- tremor of the limbs,
- insufficient coordination of movements with loss of balance,
- decreased muscle tone,
- various sleep disorders,
- frequent hysterical fits in childhood,
- speech disorders (the child starts talking late, has poor communication skills and slurred speech),
- hyperactivity against the background of increased excitability,
- difficulties concentrating and learning.
But this is a generalized picture of the disease. In fact, the clinical picture of Angelman syndrome largely depends on the stage of development of the disease and the type of chromosomal mutation that caused the pathology. This means that the symptoms of the disease can differ significantly in different patients, which for a long time did not allow us to distinguish the pathology from others with a similar clinical picture.
Among the total number of symptoms, we can highlight those that are characteristic of all patients without exception:
- severe mental retardation,
- inappropriate behavior (unreasonable laughter, increased excitability, poor concentration, state of euphoria),
- underdevelopment of motor skills,
- poor coordination of movements, gait ataxia (uneven pace, swaying from side to side, etc.), tremor of the limbs.
- speech development disorder with a predominance of non-verbal means of communication.
Among the symptoms encountered by the vast majority of patients, the following can be distinguished:
- disproportion between the head and body caused by delayed physical development,
- in many patients the shape of the skull is such that the size of the brain remains smaller than in healthy people (microcephaly),
- epileptic seizures before the age of 3 years with a progressive decrease in strength and frequency at an older age,
- distortion of EEG parameters (fluctuations and high amplitude of low-frequency waves).
These symptoms are quite common, however, 20% of patients with Angelman syndrome do not have them.
Even less frequently, it is possible to diagnose such manifestations of the disease as:
- severe or mild strabismus,
- poor control of tongue movement, resulting in patients often sticking out their tongue for no reason,
- difficulties with swallowing and sucking, especially in young children,
- disruption of skin and eye pigmentation,
- arms raised or bent while walking,
- hyperreflexia,
- sleep disorders, especially in childhood,
- frequent salivation,
- insatiable thirst,
- overly active chewing movements,
- hypersensitivity to heat,
- flat back of head,
- protruding lower jaw,
- smooth palms.
Quite a large percentage of patients have problems with urination, which they poorly control, impaired fine motor skills, which creates difficulties in self-care and learning, and excess weight. Almost all patients experience puberty later than healthy peers.
Children with Angelman syndrome perceive oral speech well and understand it, but do not want to participate in conversation, limiting their speech to several dozen words necessary in everyday life. However, in adulthood, such patients look younger than their peers without genetic pathologies.
Many symptoms of Angelman syndrome are inconstant, so the clinical picture of the disease changes significantly with age. Convulsions and epileptic seizures become less frequent or disappear altogether, the patient becomes less excitable, and sleep improves.
Complications and consequences
Angelman syndrome is a severe, currently virtually incurable chromosomal pathology that deprives patients of the opportunity to live a normal life. What the life of a child with AS will be like largely depends on the type of chromosomal abnormality.
Duplication of a chromosome segment is incompatible with life in most cases. And even if such patients do not die in infancy and reach puberty, they have no chance of having children.
The deletion or absence of a part of the genes that occurs most often in Angelman syndrome is an obstacle to the child learning to walk and talk. Such children have a more severe form of mental retardation, and epileptic seizures occur more often, and their intensity is much greater than in patients with other chromosomal abnormalities.
If there is only a mutation of one gene, with due attention and approach the child can be taught the basics of self-care, communication and interaction in a group, although he will still lag behind his peers in development.
For children with Angelman syndrome, who are kind by nature, the most important thing is the love and attention of their parents. Only in this case will the child's education bear fruit, even if small. Of course, patients with AS will not be able to study in a regular school. They need special classes where children will first be taught to concentrate, and then gradually will be given the basics of school knowledge.
Diagnostics Angelman syndrome
Angelman syndrome is a congenital developmental pathology. But due to certain circumstances, it is often impossible to diagnose it in infancy and early childhood. This is due to the non-specificity and weak expression of symptoms in infants and children under 3 years of age. And the prevalence of the disease in our country is not so great that doctors have learned to recognize it among its peers.
Angelman syndrome in infants can manifest itself as decreased muscle tone, which manifests itself in problems with feeding (weakness of the sucking and swallowing reflex), and later difficulties in learning to walk (such children begin to walk much later). These symptoms are the first signs of a developmental abnormality in the baby, which may well be associated with a chromosomal abnormality. Only genetic analysis can confirm this assumption.
Special attention is paid to children whose parents have various genomic or chromosomal disorders. After all, the disease may not manifest itself at first, and if the pathology is detected in time, by starting to work intensively with the child, it is possible to achieve significantly greater success in learning, slowing down the progression of the disease.
If the parents have various chromosomal abnormalities, genetic analysis is carried out even before the baby is born, since SA is one of the pathologies that can be detected in the embryonic stage.
The collection of material for genetic research can be carried out in two ways:
- invasive (with a certain percentage of risk, since it is necessary to penetrate the uterus in order to take a sample of amniotic fluid),
- non-invasive (analysis of the baby's DNA from the mother's blood).
The following studies are then carried out:
- fluorescent in situ hybridization (FISH method) – binding of a DNA probe labeled with a special dye to the DNA being studied, followed by examination under a microscope.
- analysis of mutations in the ube3a gene and imprinted genes,
- DNA methylation analysis using special methods used in genetics.
Genetic tests provide fairly accurate information in the case of chromosomal abnormalities, which means that future parents know in advance what to prepare for. However, there are exceptions. In a certain group of patients, in the presence of all the symptoms indicating pathology, the test results remain normal. That is, pathology can only be identified by carefully observing the child from early childhood: how he eats, when he began to walk and talk, whether he bends his legs when walking, etc.
In addition to the FISH method, among the instrumental diagnostic methods for Angelman syndrome, one can single out tomography (CT or MRI), which helps determine the condition and size of the brain, and an electroencephalogram (EEG), which shows how individual parts of the brain work.
Doctors usually make a final diagnosis at the age of 3-7 years, when the patient already has most of the symptoms and the dynamics of the disease’s development are visible.
What tests are needed?
Differential diagnosis
Angelman syndrome is a genetic pathology that has virtually no specific manifestations. Most symptoms can equally indicate both AS and other genetic pathologies.
Differential diagnosis of Angelman syndrome is carried out with the following pathologies:
- Pitt-Hopkins syndrome (patients are characterized by mental retardation, cheerful character, smiling, they have a rather large and wide mouth, microcephaly is noted). The difference is attacks of hyperventilation and breath holding in a waking state.
- Christianson syndrome (patients are mentally retarded people with a cheerful disposition, unable to speak, characterized by microcephaly, ataxia, convulsions, involuntary muscle movements).
- Mowat-Wilson syndrome (symptoms: mental retardation, epileptic seizures, pointed chin, open mouth, happy expression on the face, microcephaly). Distinction: large distance between the eyes, eyes slanted inward, rounded tip of the nose, backward-turned auricle.
- Kabuki syndrome (characterized by mild to moderate mental retardation, speech and motor problems, muscle weakness, epileptic seizures, microcephaly, long intervals between itches, and impaired coordination). Characterized by arched eyebrows, everted lateral portion of the lower eyelid, wide-set eyes, long palpebral fissures with long, thick eyelashes.
- Rett syndrome (differentiation from AS in women). Symptoms: delayed speech development, seizures, microcephaly. The difference is that there is no happy expression on the face, there are attacks of apnea and apraxia, which progresses over time.
- Autosomal recessive mental tardation syndrome 38 (symptoms: marked mental retardation with delays in motor skills and speech, muscle weakness, feeding problems in infancy, impulsivity). Distinguishing feature is the blue color of the iris.
- MECP 2 gene duplication syndrome (differentiation from SA in males). Symptoms: severe mental retardation, muscle weakness since childhood, speech problems or lack of speech, epilepsy. Distinctions: progressive myopathy, constantly recurring infections.
- Kleefstra syndrome (symptoms: speech and thinking problems, muscle weakness, sleep disturbances, lack of attention, open mouth, hyperactivity, seizures, ataxia, balance disorders). Distinctive features: flat face, short snub nose, wide-set eyes, large everted lower lip, aggressive outbursts.
- Smith-Magenis syndrome (characterized by seizures, sleep problems, intellectual and motor development disorders). Distinctive features include a broad and flat face and a prominent forehead.
- Koolen-de Vries syndrome (mild to moderate mental retardation, muscle weakness, seizures, friendliness). Distinguishing features: long face with high forehead, protruding ears, slanted eyes, high joint mobility, congenital heart defects.
- Phelan-McDermid syndrome (symptoms: mental retardation, speech disorders or lack of speech). Distinctions: large hands with developed muscles, muscle weakness from birth, weak sweating.
Such pathologies as adenyl succinate deficiency, autosomal recessive mental retardation syndrome 1, chromosome 2q23.1 duplication syndrome, FOXG1, STXBP1 or MEF2C gene haploinsufficiency syndromes and some others can “boast” of symptoms similar to Angelman syndrome.
The doctor's task is to make an accurate diagnosis, differentiating Angelman syndrome from pathologies with similar symptoms, and prescribe effective treatment that is relevant to the diagnosed stage of the disease.
Who to contact?
Treatment Angelman syndrome
Angelman syndrome is one of those pathologies for which medicine is still searching for effective treatment. The etiological treatment of the disease is in the development stage of various methods and means, many of which have not yet been tested on humans. This means that for now doctors have to limit themselves to symptomatic therapy, which helps to somehow alleviate the unenviable situation of children and adults with marionette syndrome, suffering from epileptic seizures, salivation, hypotension and sleep disorders.
Thus, it is possible to reduce the frequency and strength of epileptic seizures with the help of a properly selected anticonvulsant drug. But the whole difficulty is that seizures in patients with SA differ from ordinary epileptic seizures in that they are characterized by several types of seizures, which means that the condition can be alleviated by administering several drugs at once.
The most popular anticonvulsants used to treat Angelman syndrome are: valproic acid, topiramate, lamotrigine, levetiracetam, clonazepam and drugs based on them. Less commonly used are drugs based on carmazepine, phenytoin, phenobarbital, ethosuximide, since some of them can provoke a paradoxical effect consisting in strengthening and increasing the frequency of epileptic seizures. This happens if the drug is used as part of monotherapy.
To treat drooling, two methods are usually used: medicinal (drugs that suppress saliva production) and surgical, which involves reimplantation of the salivary ducts. But in the case of SA, these methods are considered ineffective, and the issue remains open. Parents and those who care for such patients have to pay special attention to this issue, since the patients themselves usually do not control drooling, and some are simply unable to take care of themselves.
Another problem is short sleep duration. Often children with Angelman syndrome sleep no more than 5 hours, which has a negative impact on the functioning of the entire body. Easily excitable, active children who love games and communication (even if they try to limit themselves to non-verbal methods) are noticeably tired during the day. In order to have a good rest, the body needs a deep, full sleep, but this is precisely the catch.
It would seem that sedative drugs (phenothiazines and atypical antipsychotics) that calm the nervous system should be sufficient to improve sleep in excitable patients. But in the case of AS, the use of such drugs is fraught with the occurrence of negative effects. Therefore, doctors still prefer mild sleeping pills, such as Melatonin (a natural hormonal drug based on the sleep hormone), which is given to patients an hour before going to bed in the amount of 1 tablet, and Diphenhydramine. The frequency of administration and dosage of which is determined by the doctor depending on the condition and age of the patient.
Sometimes patients with Angelman syndrome have problems with digestion and stool. You can improve your stool with laxatives (preferably herbal ones).
Or you can approach the problem differently, as American doctors did, based on some methods of treating autism, because many symptoms characteristic of AS are also characteristic of autism (impulsivity, involuntary movements, repetitive actions, attention deficit, communication problems, etc.). It was noted that the introduction of the hormone secretin, which normalizes digestion and stool, has a positive effect on the attention of patients, and oxytocin helps improve the child's cognitive abilities and memory, and correct behavior.
True, hormones alone are not enough, especially when it comes to children. In Angelman syndrome, behavioral therapy, work with a psychologist and speech therapist (teaching non-verbal communication methods and sign language) are indicated. The education of such children should be based on an individual program with the participation of specially trained teachers, a psychologist and parents. Unfortunately, this is not possible everywhere, and families are left alone with their problem.
Since many young patients with AS suffer from low muscle tone and joint problems, much attention is paid to physiotherapy. Most often, doctors resort to the use of paraffin applications, electrophoresis, and magnetic therapy.
Active tonic massage and special exercises of therapeutic physical training will help the sick child to stand on his feet and walk confidently after a while. Aquagymnastics is especially useful in this regard, which is recommended for SA in cool water. It increases muscle tone and teaches the child to control his body and coordinate movements.
Anticonvulsant treatment
The most dangerous symptom of Angelman syndrome is seizures similar to those of epilepsy. This symptom is observed in 80% of patients, which means that all of them need to be prescribed effective anticonvulsant treatment.
Treatment of epileptic seizures is carried out with the help of vitamins and anticonvulsants. In Angelman syndrome, accompanied by a convulsive syndrome, vitamins of group B, as well as vitamins C, D and E will be useful. But prescribing vitamin therapy on your own in this case is very dangerous, because uncontrolled intake of vitamins can reduce the effectiveness of antiepileptic drugs and provoke new, more severe and prolonged seizures.
The selection of anticonvulsant drugs and the prescription of their effective dosage should also be done by a specialist doctor. He or she also decides whether one drug will be enough or whether the patient will have to take 2 or more drugs for a long time.
For most patients, doctors prescribe valproic acid drugs (Valproic acid, Depakine, Convulex, Valparin, etc.), which prevent seizures and improve the mood and mental state of patients.
Valproic acid is available in the form of tablets, syrup and injection solutions. The most popular drug is the prolonged-release drug "Depakine" in tablets and as a solution for intravenous administration. The dosage of the drug is determined by the doctor individually depending on the weight, age and condition of the patient.
The drug is taken during meals 2 to 3 times a day. The average daily dose is 20-30 mg per 1 kilogram of the patient's weight, the maximum is 50 mg/kg per day.
Contraindications for use. Do not use in case of liver and pancreas dysfunction, hemorrhagic diathesis, hepatitis, porphyria and hypersensitivity to the drug.
Side effects include hand tremors, digestive and stool disorders, and changes in body weight.
"Topiramate" is also a drug of choice for SA. It is produced in tablet form and is used both as part of monotherapy and in combination with other drugs.
Method of administration and dosage. Take the tablets orally regardless of food intake. The initial daily dose for adults is 25-50 mg, for children - 0.5-1 mg/kg. Each week, the dosage is increased according to the doctor's instructions.
The drug should not be taken during pregnancy and lactation, as well as in case of hypersensitivity to its components. The drug has many different side effects.
Drugs that a doctor may prescribe for Angelman syndrome: Clomazepam, Rivotril, Lamotrigine, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra, etc.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Traditional medicine and homeopathy
Traditional medicine, like homeopathic preparations, are of course relatively safe, but the effectiveness of such treatment for Angelman syndrome can be considered controversial.
Although folk treatment can still help in some things. We are talking about stopping epileptic seizures. In this regard, herbal treatment can be quite effective.
A good effect is provided by a medicinal collection based on peony, licorice and duckweed (the components are taken in equal quantities). The herbs need to be ground into flour. After 2 weeks from the start of taking it, you can notice a significant decrease in the frequency of seizures.
Lavender decoction (1 teaspoon per glass of boiling water) is also useful for cramps. The mixture is boiled for 5 minutes and infused for half an hour. The medicine is taken at night for 14 days.
An aqueous (or alcoholic) infusion of motherwort is considered effective for epileptic seizures.
Of the homeopathic preparations for preventing seizures in Angelman syndrome, you can use medicines based on chamomile and motherwort, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. But it should be taken into account that only a homeopathic doctor can prescribe effective and safe doses of drugs in each specific case.
Prevention
As the reader has probably already understood, medicine is not yet able to prevent gene mutations and other chromosomal abnormalities, however, as well as to correct the situation. This can happen to anyone, because children with Angelman syndrome are born to healthy parents, and genetics, which is currently one of the least studied branches of medicine, cannot yet explain this.
The only thing that can be done is to take a responsible approach to pregnancy planning, register and undergo examinations in time. But again, such a measure will be more educational than preventive, like any examinations. But young parents will know in advance what to prepare for, and in case of a positive answer, they will decide whether they can take on such responsibility as raising a sick child.
Forecast
The prognosis for Angelman syndrome depends on the nature of the chromosomal abnormality and the timeliness of its detection. The hardest hit are those children whose chromosome 15 contains "gaps" in genes (deletion). The likelihood of such patients walking and talking is extremely low. Other cases can be corrected with a careful approach and love for your child.
Unfortunately, such patients will not be able to become full-fledged members of society, despite the fact that they are far from stupid, they understand speech and its meaning. However, they will have problems with communication for the rest of their lives. Patients can be taught sign language from childhood, but they cannot be forced to communicate using words. The vocabulary of "speaking" patients is limited to the minimum of words used in everyday life (5-15 words).
As for the life expectancy and general health of patients with Angelman syndrome, the figures here fluctuate around average values. In adulthood, patients mostly face health problems such as scoliosis and obesity, which, with the right approach to treatment, are not life-threatening.