Medical expert of the article

New publications
Achondroplasia
Last reviewed: 12.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
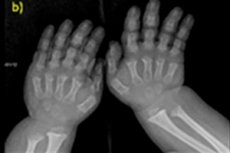
There are many rare congenital diseases, and one of them is a violation of bone growth - achondroplasia, which leads to severe disproportionate short stature.
In the section on developmental anomalies of ICD-10, the code for this type of hereditary osteochondral dysplasia with growth defects of tubular bones and spine is Q77.4 [ 1 ]
Epidemiology
Regarding the prevalence of achondroplasia, statistical data from various studies are ambiguous. Some claim that this anomaly occurs in one newborn out of 10 thousand, others - in one out of 26-28 thousand, and still others - 4-15 cases out of 100 thousand. [ 2 ]
There is also information that when the father is over 50 years old, the incidence of achondroplasia in children is one case per 1875 newborns.
Causes achondroplasia
The cause of achondroplasia is a violation of osteogenesis, in particular, one of the types of intrauterine ossification of the diaphyses of the tubular bones of the skeleton - endochondral ossification, during which cartilage is modified into bone tissue. For more details, see - Bone development and growth
Disruption of ossification of long bones, i.e. fetal achondroplasia, occurs due to mutations in the membrane tyrosine kinase gene - fibroblast growth factor receptor 3 (FGFR3 on chromosome 4p16.3), which affects cell growth and differentiation. The presence of FGFR3 mutations is associated with genetic instability and changes in the number of chromosomes (aneuploidy).
Achondroplasia is transmitted to a child as an autosomal dominant trait, that is, he receives one copy of the mutant gene (which is dominant) and one normal gene on a pair of non-sex (autosomal) chromosomes. Thus, the type of inheritance of this defect is autosomal dominant, and the anomaly can manifest itself in 50% of offspring when a combination of alleles of this gene (genotype) is crossed.
In addition, mutations can be sporadic, and, as practice shows, in 80% of cases children with achondroplasia are born to parents of normal height.
Risk factors
The main risk factors for the birth of children with achondroplasia are hereditary. If one of the parents has this defect, then the probability of having a sick child is estimated at 50%; if both parents have this anomaly, it is also 50%, but with a 25% risk of homozygous achondroplasia, leading to death before birth or in early infancy.
With the age of the father (closer to 40 years and older), the risk of a new mutation (de novo mutation) of the FGFR3 gene increases.
Pathogenesis
Explaining the pathogenesis of achondroplasia, experts emphasize the importance of the transmembrane protein tyrosine protein kinase (encoded by the FGFR3 gene) in regulating the division, differentiation and apoptosis of cells of the cartilage tissue of the growth plates - chondrocytes, as well as the normal development of the skeleton - osteogenesis and mineralization of bone tissue.
During embryonic development, in the presence of a gene mutation, the receptors of fibroblast growth factor 3 become more active. The increase in their functions disrupts the transmission of cellular signals and the interaction of the extracellular part of this protein with polypeptide fibroblast growth factors (FGF). As a result, a failure occurs: the stage of proliferation of cartilage cells becomes shorter, and their differentiation begins earlier than expected. All this leads to improper formation and fusion of the skull bones and skeletal dysplasia - a decrease in long bones, which is accompanied by pronounced short stature or dwarfism.
And two thirds of cases of dwarfism are associated with achondroplasia.
Symptoms achondroplasia
Abnormal bone growth causes clinical symptoms of achondroplasia such as:
- pronounced short stature (disproportionate dwarfism) with an average adult height of 123-134 cm;
- shortening of the proximal parts of the lower and upper limbs with a relatively normal torso size;
- shortened fingers and toes;
- enlarged head (macro or megalocephaly); [ 3 ]
- specific facial features in the form of a protruding forehead and hypoplasia of the middle part of the face - a sunken bridge of the nose.
- narrow craniocervical junction. Some infants with achondroplasia die in the first year of life from complications related to the craniocervical junction; population studies suggest that this excess risk of death may be as high as 7.5% without evaluation and intervention.[ 4 ]
- Middle ear dysfunction is often a problem [ 5 ], and if not treated properly can lead to conductive hearing loss severe enough to interfere with speech development. More than half of children will require a pressure equalization tube. [ 6 ] Overall, about 40% of people with achondroplasia have functionally significant hearing loss. Expressive language development is also often delayed, although the strength of the relationship between hearing loss and expressive language problems is questionable.
- Bowing of the tibia is very common in people with achondroplasia. Over 90% of untreated adults have some degree of bowing.[ 7 ] "Bowing" is actually a complex deformity resulting from a combination of lateral tilt, internal torsion of the tibia, and dynamic instability of the knee.[ 8 ]
Infants with achondroplasia are characterized by muscular hypotonia, due to which they begin to learn movement skills and walk later. Intelligence and cognitive abilities are not affected by this developmental defect. [ 9 ], [ 10 ]
Consequences and complications
This type of hereditary osteochondral dysplasia is characterized by the following complications and consequences:
- recurrent ear infections;
- obstructive sleep apnea;
- hydrocephalus;
- malocclusion and crooked teeth:
- deformation of the legs (varus or valgus) with a change in gait;
- hypertrophied lordosis of the lumbar spine or its curvature (thoracolumbar kyphosis or lumbar scoliosis) - with back pain when walking;
- joint pain (due to incorrect positioning of bones or compression of nerve roots);
- Spinal stenosis and spinal cord compression; The most common medical complaint in adulthood is symptomatic spinal stenosis involving L1-L4. Symptoms range from intermittent, reversible claudication induced by exercise to severe, irreversible leg dysfunction and urinary retention.[ 11 ] Claudication and stenosis can cause both sensory (numbness, pain, heaviness) and motor symptoms (weakness, stumbling, limited walking endurance). Vascular claudication results from swelling of blood vessels after standing and walking and is completely reversible with rest. Spinal stenosis is the actual lesion of the spinal cord or nerve root by the stenotic bone of the spinal canal, and the symptoms are irreversible. Symptoms localized to a particular dermatome may result from stenosis of specific nerve root foramina.
- a reduction in the chest wall with limited lung growth and decreased lung function (severe shortness of breath). In infancy, a small group of people with achondroplasia have restrictive lung problems. Small breasts and increased chest compliance combine to result in reduced lung capacity and restrictive lung disease [ 12 ]
Other orthopedic problems
- Joint weakness. Most joints are hypermobile in childhood. Generally, this has little effect, except for knee instability in some people.
- Discoid lateral meniscus: This recently identified structural abnormality may lead to chronic knee pain in some people.[ 13 ]
- Arthritis: Constitutive activation of FGFR-3, as in achondroplasia, may protect against the development of arthritis.[ 14 ]
- Acanthosis nigricans is seen in approximately 10% of people with achondroplasia.[ 15 ] In this population, it does not reflect hyperinsulinemia or malignancy.
Homozygous achondroplasia caused by biallelic pathogenic variants at nucleotide 1138 of FGFR3 is a severe disorder with radiologic findings qualitatively different from those seen in achondroplasia. Early death results from respiratory failure due to a small chest wall and neurologic deficits due to cervicomedullary stenosis [Hall 1988].
Diagnostics achondroplasia
In most patients, the diagnosis of achondroplasia is made based on characteristic clinical signs and radiographic findings. In infants or in the absence of some symptoms, genetic testing, such as karyotype analysis, is used to make a definitive diagnosis.[ 16 ]
When performing prenatal diagnostics using the molecular genetics method, analyses of amniotic fluid or a chorionic villus sample can be performed.
Signs of achondroplasia on ultrasound of the fetus - shortening of the limbs and typical facial features - are visualized after 22 weeks of pregnancy.
Instrumental diagnostics also includes X-ray of the skeleton or ultrasound of the bones. And X-ray confirms the diagnosis based on such data as a large skull with a narrow occipital foramen and a relatively small base; short tubular bones and shortened ribs; short and flattened vertebral bodies; narrowed spinal canal, reduced size of the iliac wings.
Differential diagnosis
Differential diagnostics with pituitary dwarfism, congenital spondyloepiphyseal and diastrophic dysplasia, hypochondroplasia, Shereshevsky-Turner and Noonan syndromes, pseudoachondroplasia are necessary. Thus, the difference between pseudoachondroplasia and achondroplasia is that in patients with dwarfism in pseudoachondroplasia, the head size and facial features are normal.
Who to contact?
Treatment achondroplasia
Recommendations for the care of children with achondroplasia have been outlined by the American Academy of Pediatrics Committee on Genetics. These recommendations are intended to provide guidance and are not intended to replace individual decision-making. A recent review [Pauli & Botto 2020] also includes guidelines. There are specialty clinics that specialize in the treatment of skeletal dysplasia; their recommendations may differ slightly from these general recommendations.
Recommendations include (but are not limited to) the following.
Hydrocephalus. If signs or symptoms of increased intracranial pressure appear (e.g., accelerated head growth, persistently bulging fontanelle, noticeable enlargement of superficial veins on the face, irritability, vomiting, vision changes, headache), referral to a neurosurgeon is necessary.
The presumed etiology of hydrocephalus in achondroplasia is increased intracranial venous pressure due to stenosis of the jugular foramina. Therefore, the standard treatment has been ventriculoperitoneal shunting. However, endoscopic third ventriculostomy may be beneficial in some individuals,[ 17 ] implying that other mechanisms, such as fourth ventricular outlet obstruction due to craniocervical stenosis, may be involved.[ 18 ]
Craniocervical junction stenosis. Best predictors of need for suboccipital decompression:
- Hyperreflexia or clonus of the lower extremities
- Central hypopnea on polysomnography
- Reduction in foramen magnum size as determined by computed tomography of the craniocervical junction and compared with norms for children with achondroplasia.[ 19 ]
- Evidence of spinal cord compression and/or T2-weighted signal abnormalities have recently been suggested as another factor to consider when deciding to operate.
If there are clear signs of symptomatic compression, an urgent referral to a pediatric neurosurgeon should be made for decompression surgery. [ 20 ]
Obstructive sleep apnea Treatment may include:
- Adenotonsillectomy
- Positive airway pressure
- Tracheostomy in extreme cases
- Weight loss
These interventions may result in improvement in sleep disturbance and some improvement in neurological function.[ 21 ]
In rare cases where obstruction is severe enough to require tracheostomy, midface advancement surgery has been used to relieve upper airway obstruction.[ 22 ]
Middle ear dysfunction. Frequent middle ear infections, persistent middle ear fluid, and subsequent hearing loss should be treated aggressively when needed. Long-term tubes are recommended because they are often needed until age seven or eight.[ 23 ]
When problems arise at any age, it is recommended to use appropriate treatment methods.
Short stature. Several studies have evaluated growth hormone (GH) therapy as a possible treatment for short stature achondroplasia.[ 24 ]
Overall, these and other series show an initial acceleration of growth, but the effect diminishes over time.
On average, you can expect an increase in adult height of only about 3 cm.
Extended limb lengthening using various techniques remains an option for some. Height gains of up to 30-35 cm can be achieved. [ 25 ] Complications are common and can be serious.
While some advocate performing these procedures as early as six to eight years of age, many pediatricians, clinical geneticists, and ethicists advocate delaying such surgery until a young person is able to participate in making an informed decision.
At least in North America, only a small proportion of affected individuals choose to undergo advanced limb lengthening. The Little People of America Medical Advisory Board has issued a statement regarding the use of advanced limb lengthening.
Obesity: Measures to prevent obesity should begin in early childhood. Standard treatments for obesity should be effective in people with achondroplasia, although caloric needs are lower. [ 26 ]
Standard weight and weight-for-height charts specific to achondroplasia should be used to track progress. It is important to note that these curves are not perfect weight-for-height curves; they were derived from thousands of data points from people with achondroplasia.
Body mass index (BMI) standards were developed for children aged 16 years and younger. [ 27 ] BMI is not standardized for adults with achondroplasia; comparisons to BMI curves for average height will give misleading results. [ 28 ]
Varus deformity. Annual orthopedic follow-up by either a provider familiar with achondroplasia or an orthopedic surgeon is recommended. Criteria for surgical intervention have been published.[ 29 ]
The presence of progressive symptomatic curve requires referral to an orthopaedist. Asymptomatic varus deformity itself does not usually require surgical correction. Various interventions may be chosen (eg, guided growth using eight plates, valgus osteotomy, and derotational osteotomy). There are no controlled studies comparing the results of treatment options.
Kyphosis. Infants with achondroplasia often develop a flexible kyphosis. A protocol is available to help prevent the development of a fixed angular kyphosis, which includes avoiding flexible strollers, swings, and baby carriers. Advice against unsupported sitting; always apply counterpressure to the back when holding the baby.
- Kyphosis improves significantly or resolves in most children after adopting an orthograde posture and starting to walk. [ 30 ]
- In children who do not spontaneously remit after increasing trunk strength and starting to walk, bracing is usually sufficient to prevent persistence of the thoracolumbar kyphosis.[ 31 ]
- If severe kyphosis persists, spinal surgery may be required to prevent neurological complications.[ 32 ]
Spinal Stenosis: If severe signs and/or symptoms of spinal stenosis occur, urgent referral to a surgical specialist is necessary.
Extended and wide laminectomy are usually recommended. The relevance of the procedure depends on the level (e.g. thoracic or lumbar) and the degree of stenosis. Patients had better outcomes and improved function the sooner they had surgery after the onset of symptoms [ 33 ]
Immunizations: Nothing about achondroplasia precludes all routine immunizations. Given the increased respiratory risk, DTaP, pneumococcal, and influenza vaccines are especially important.
Adaptive Needs: Because of short stature, environmental modifications may be necessary. In school, this may include stools, lowered light switches, toilets of appropriate height or other means of accessibility, lower desks, and footrests in front of chairs. All children should be able to exit the building independently in the event of an emergency. Small hands and weak tendons may make fine motor skills difficult. Appropriate accommodations include using a smaller keyboard, weighted pens, and smoother writing surfaces. Most children should have an IEP or 504 plan.
Pedal extensions are almost always needed for riding. Workstation modifications such as lower tables, smaller keyboards, steps, and toilet access may also be required.
Socialization: Because of the very noticeable short stature associated with achondroplasia, affected individuals and their families may have difficulty socializing and adjusting to school.
Support groups such as Little People of America, Inc (LPA) can help families address these issues through peer support, personal example, and social awareness programs.
Information on employment, education, disability rights, adoption of short children, medical issues, appropriate clothing, adaptive devices and parenting is available through a national newsletter, seminars and workshops.
There is no medicine or non-drug treatment that can cure this congenital defect.
Physical therapy is most commonly used; treatment may also be needed for hydrocephalus (by shunt or endoscopic ventriculostomy), obesity, [ 34 ] apnea, [ 35 ] middle ear infection, or spinal stenosis.
In some clinics, after the child reaches five to seven years of age, they undertake surgical treatment: lengthening the bones of the shins, thighs and even shoulder bones or correcting the deformity - with the help of operations and special orthopedic devices - in three to four stages, each lasting up to 6-12 months.
Therapy under investigation
The administration of a C-type natriuretic peptide analogue is undergoing clinical trials. Initial results have shown that it is well tolerated and results in an increase in growth velocity from baseline in children with achondroplasia ( trial site ). [ 36 ] Conjugated C-type natriuretic peptide is also currently undergoing clinical trials. [ 37 ] Other considerations include tyrosine kinase inhibition [ 38 ], meclizine [ 39 ], and a soluble recombinant human FGFR3 decoy. [ 40 ]
Search clinicaltrials.gov in the US and the EU Clinical Trials Registry in Europe for information on clinical trials for a wide range of diseases and conditions.
Prevention
The only preventive measure is prenatal diagnosis of congenital diseases. [ 41 ], [ 42 ]
Forecast
How long do people with achondroplasia live? About 10 years less than the average life expectancy.
Since pathological changes in bone tissue and joints lead to limitations of self-care and mobility, children with this diagnosis are given the status of disabled. In the long term, most patients have a normal prognosis, but with age, there is an increased risk of heart disease. [ 43 ]