Medical expert of the article

New publications
Hemolytic jaundice: causes, symptoms, diagnosis and treatment
Last updated: 17.04.2026

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
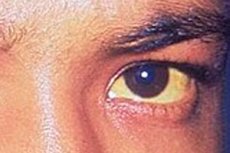
Hemolytic jaundice is a type of jaundice that occurs due to the rapid destruction of red blood cells. When hemoglobin breaks down faster than the liver can capture, conjugate, and eliminate bilirubin, predominantly unconjugated, or indirect, bilirubin begins to accumulate in the blood. This mechanism underlies so-called prehepatic jaundice. [1] [2]
From a clinical perspective, hemolytic jaundice is not a distinct entity. It is a syndrome that can occur in a wide variety of diseases, from hereditary spherocytosis and glucose-6-phosphate dehydrogenase deficiency to autoimmune hemolytic anemia, sickle cell disease, malaria, thrombotic microangiopathies, and paroxysmal nocturnal hemoglobinuria. Therefore, in practice, it is not "jaundice in general" that is treated, but the specific cause of hemolysis. [3] [4] [5] [6]
Unlike mechanical jaundice, the hemolytic form usually does not have a primary bile duct block. Unlike hepatic jaundice, the problem does not begin with damage to hepatocytes. The main pathogenic node is located earlier – in the blood and the red blood cell destruction system, although with severe and prolonged hemolysis, the liver and bile ducts also become secondarily involved. [7] [8]
There are two key things for patients to understand. First, a yellow tint to the skin and sclera associated with hemolytic jaundice may not be the most severe manifestation of the disease: sometimes, anemia itself, hemoglobinuria, thrombosis, renal damage, or a hemolytic crisis are more dangerous. Second, modern diagnostics allow for the rapid differentiation of hemolytic jaundice from other types by immediately assessing bilirubin fractions, reticulocytes, lactate dehydrogenase, haptoglobin, a blood smear, and a direct antiglobulin test. [9] [10] [11]
| What is important to know right away | Practical meaning |
|---|---|
| Hemolytic jaundice is a syndrome | It is necessary to look for the cause of hemolysis |
| The main type of bilirubin | Indirect bilirubin usually increases |
| This is a prehepatic form of jaundice. | The main problem starts before the liver |
| Common causes | Hereditary hemolytic anemia, autoimmune hemolysis, microangiopathy, infections |
| The main principle of treatment | Treat the source of hemolysis, not just the jaundice itself. |
Source of the table. [12] [13]
Code according to ICD 10 and ICD 11
There is no single universal code for adult hemolytic jaundice that completely replaces the cause code. In the International Classification of Diseases, 10th revision, such cases are typically coded using the underlying hemolytic disease from the range D55-D59, which encompasses hemolytic anemias. If the symptom of jaundice is present without a definitive explanation, the World Health Organization also uses the category R17.0 - hyperbilirubinemia with mention of jaundice, not elsewhere classified, while a number of clinical modifications use R17 - unspecified jaundice. [14] [15]
In practice, this means that in cases of hereditary spherocytosis, the code is for hereditary hemolytic anemia, while in cases of autoimmune hemolytic anemia, the corresponding code is for acquired hemolytic anemia, and jaundice itself may be a manifestation. This approach is more accurate because it reflects the pathogenesis: jaundice here is secondary to hemolysis. [16] [17]
The International Classification of Diseases, 11th revision, follows the same logic. There is no separate code for "hemolytic jaundice" as a distinct adult disorder, and the primary coding is built around block 3A10-3A4Z - hemolytic anemias. For hereditary forms, section 3A10 is used, for acquired forms, the more detailed codes of block 3A20 and further are used, and the symptom of jaundice, if necessary, can be reflected by code ME10.1 - unspecified jaundice. [18] [19] [20]
Therefore, in an article on hemolytic jaundice, it is more appropriate to speak not of a single code, but of a coding approach. First, the specific hemolytic anemia is recorded, then, if necessary for documentation, the jaundice itself is reflected as a manifestation of hyperbilirubinemia. This is how the coding remains clinically meaningful. [21] [22]
| Clinical situation | International Classification of Diseases, 10th revision | International Classification of Diseases, 11th revision |
|---|---|---|
| Hemolytic anemia as a cause of jaundice | D55-D59 | 3A10-3A4Z |
| Hereditary hemolytic anemia | D58 and related headings | 3A10 |
| Acquired hemolytic anemia | D59 and related headings | 3A20 and related codes |
| Jaundice as a symptom without a clear cause | R17.0 or R17 in clinical modifications | ME10.1 |
Source of the table. [23] [24] [25] [26]
Epidemiology
Hemolytic jaundice does not have a single global prevalence because it is a syndrome encompassing many different diseases. Therefore, its epidemiology is more accurately assessed through the underlying hemolytic conditions that most often lead to jaundice. These include both rare diseases, such as autoimmune hemolytic anemia, and very common hereditary conditions, such as glucose-6-phosphate dehydrogenase deficiency. [27] [28]
Hereditary spherocytosis is considered the most common inherited hemolytic anemia associated with a red blood cell membrane defect. According to the NCBI Bookshelf, it is diagnosed in approximately 1 in 2,000 people, most commonly in Northern Europe and North America, although it also occurs in other populations. It is in these patients that the classic triad of anemia, splenomegaly, and intermittent jaundice remains very characteristic. [29]
Glucose-6-phosphate dehydrogenase deficiency is one of the most common human enzymopathies. A 2025 StatPearls review estimates that it affects approximately 400 million people worldwide, with the highest prevalence in sub-Saharan Africa, the Mediterranean, the Middle East, and South and Southeast Asia. In these regions, hemolytic jaundice often becomes part of the clinical picture following infection, medication intake, or exposure to other oxidative triggers. [30]
Sickle cell disease is another major contributor to the global epidemiology of hemolytic jaundice. The World Health Organization reports that approximately 7.74 million people were living with the disease in 2021, with the highest proportion of cases occurring in sub-Saharan Africa. Because sickle cell disease is associated with chronic hemolysis, episodes of jaundice and bilirubin overload are extremely common in these patients. [31]
Autoimmune hemolytic anemia, on the other hand, is a rare disorder, but is clinically extremely important because it can rapidly lead to severe hemolysis and jaundice. Modern epidemiological reviews from 2025 estimate the incidence of autoimmune hemolytic anemia to be between 1.4 and 6.6 cases per 100,000 people, depending on the database and diagnostic approach. This makes it rare in terms of population frequency, but highly significant for inpatient hematology. [32]
| The main cause of hemolytic jaundice | Epidemiological characteristics |
|---|---|
| Hereditary spherocytosis | About 1 case per 2000 people in some populations |
| Glucose-6-phosphate dehydrogenase deficiency | About 400 million people in the world |
| Sickle cell disease | About 7.74 million people worldwide in 2021 |
| Autoimmune hemolytic anemia | A rare disease, with an incidence of approximately 1.4-6.6 per 100,000 |
| Thrombotic microangiopathies and paroxysmal nocturnal hemoglobinuria | Much less common, but often more severe in terms of flow |
Source of the table. [33] [34] [35] [36]
Reasons
The causes of hemolytic jaundice are conveniently divided into congenital and acquired. Congenital causes include erythrocyte membrane defects, enzymopathy, and hemoglobinopathies. Acquired causes include autoimmune processes, infections, toxins, medications, mechanical damage to erythrocytes, thrombotic microangiopathy, and paroxysmal nocturnal hemoglobinuria. This approach helps the physician immediately develop a diagnostic algorithm. [37]
Among congenital causes, hereditary spherocytosis, glucose-6-phosphate dehydrogenase deficiency, and sickle cell disease are particularly important. In hereditary spherocytosis, red blood cells lose their normal shape and are destroyed more quickly in the spleen. In glucose-6-phosphate dehydrogenase deficiency, cells become vulnerable to oxidative stress. In sickle cell disease, red blood cells are deformed, have a shorter lifespan, and hemolysis becomes chronic. [38] [39] [40]
Among the acquired causes, autoimmune hemolytic anemia remains one of the most important. Merck notes that autoantibodies can destroy red blood cells at body temperature, as in the warm form, or at lower temperatures, as in cold agglutinin disease. This destruction often results in anemia, jaundice, enlarged spleen, and sometimes severe hemolytic crisis. [41]
Another major group is infections. Hemolytic jaundice can occur with malaria, babesiosis, severe bacterial infections, and some viral processes. In malaria, the rupture of infected red blood cells and their increased elimination in the spleen can cause anemia and jaundice, especially in severe forms of the infection. [42] [43]
One must also consider mechanical hemolysis, which develops in prosthetic heart valves, with severe valvular stenosis, in thrombotic microangiopathies, and in disseminated intravascular coagulation syndrome. Here, jaundice occurs as part of intravascular hemolysis, and the clinical picture is often accompanied by hemoglobinuria, schistocytes, and more pronounced systemic stress. [44] [45]
| Group of reasons | Examples |
|---|---|
| Hereditary membrane defects | Hereditary spherocytosis, hereditary elliptocytosis |
| Hereditary enzyme deficiencies | Glucose-6-phosphate dehydrogenase deficiency |
| Hemoglobinopathies | Sickle cell disease, thalassemia |
| Immune causes | Warm and cold autoimmune hemolytic anemia |
| Infectious causes | Malaria, babesiosis, severe bacterial infections |
| Mechanical and microangiopathic causes | Prosthetic valves, thrombotic microangiopathy, disseminated intravascular coagulation syndrome |
| Acquired clonal causes | Paroxysmal nocturnal hemoglobinuria |
Source of the table. [46] [47] [48] [49]
Risk factors
Risk factors depend on the specific cause of hemolysis. For hereditary forms, the main risk factors remain family history and belonging to populations where certain mutations are more common. For example, hereditary spherocytosis is more often described in Northern European populations, and glucose-6-phosphate dehydrogenase deficiency is particularly common in regions of the historical malaria belt. [50] [51]
Risk factors for autoimmune hemolytic anemia include systemic autoimmune diseases, lymphoproliferative disorders, and certain medications. Merck notes that secondary warm autoimmune hemolytic anemia can develop in the context of systemic lupus erythematosus, lymphoma, chronic lymphocytic leukemia, and after taking certain medications, including certain antibiotics and other drugs. [52]
For oxidative hemolysis in glucose-6-phosphate dehydrogenase deficiency, key triggers include infections, certain medications, and, most commonly, fava beans. Patients may not experience any noticeable symptoms for a long time until they encounter a trigger. Therefore, risk is determined not only by genetics but also by exposure to triggering factors. [53] [54]
Mechanical risk factors include prosthetic heart valves, extracorporeal devices, and significant vascular and valvular turbulence, while microangiopathic risk factors include pregnancy, malignant hypertension, severe infections, autoimmune diseases, transplantation, and cancer. Thus, risk factors can be both genetic and acutely acquired. [55] [56]
Long-term chronic hemolysis itself creates additional risks of complications. Due to persistent bilirubin overload, such patients are more likely to develop pigment gallstones. The NIDDK and Merck indicate that black pigment stones are associated with bilirubin and are more common in people with hemolytic anemia. [57] [58]
| Risk factor | For what reasons is it especially important? |
|---|---|
| Family history | Hereditary spherocytosis, hemoglobinopathies, enzymopathies |
| Ethnic and geographical origin | Glucose-6-phosphate dehydrogenase deficiency, sickle cell disease |
| Autoimmune diseases and lymphoproliferations | Autoimmune hemolytic anemia |
| Drug triggers | Oxidative hemolysis, drug-induced immune hemolysis |
| Prosthetic valves and devices | Mechanical hemolysis |
| Chronic hemolysis | Pigment gallstones and biliary complications |
Source of the table. [59] [60] [61]
Pathogenesis
The pathogenesis of hemolytic jaundice begins with accelerated destruction of red blood cells. Normally, a red blood cell lives for about 120 days, after which it is discarded. During hemolysis, its lifespan is shortened, hemoglobin breaks down more rapidly, and the flow of bilirubin to the liver increases sharply. If the liver is unable to bind and remove this entire volume, indirect bilirubin accumulates in the blood. [62] [63]
Hemolysis can be extravascular or intravascular. In the extravascular variant, red blood cells are most often destroyed in the spleen and liver, which is typical, for example, of hereditary spherocytosis and many variants of autoimmune hemolytic anemia. In intravascular hemolysis, cells are destroyed directly in the bloodstream, which is most often observed in paroxysmal nocturnal hemoglobinuria, microangiopathies, severe mechanical hemolysis, and some toxic conditions. [64] [65]
In hemolytic jaundice, indirect bilirubin is predominantly elevated. This results in more bilirubin metabolites entering the intestines, so stool often remains normal-colored or even darker than usual due to elevated stercobilin, while urobilinogen in the urine may increase. Unlike cholestatic jaundice, the typical sudden lightening of stool is usually absent. [66] [67]
If hemolysis continues for a long time, the bilirubin load alters the composition of bile. This leads to the more frequent formation of black pigment stones, which consist of bilirubin compounds. This is why chronic hemolytic anemias are often complicated by cholelithiasis, even if the liver itself is initially structurally intact. [68] [69]
In the most severe forms of hemolysis, other organs are also involved in the pathogenesis. Anemic hypoxia, hemoglobinuria, splenic overload, risk of thrombosis, and, in the case of intravascular hemolysis, kidney damage appear. Therefore, hemolytic jaundice should always be considered as part of a systemic process, and not as an isolated pigment problem. [70] [71] [72]
| Stage of pathogenesis | What's happening |
|---|---|
| Accelerated destruction of red blood cells | The flow of bilirubin to the liver increases |
| Conjugation system overload | Indirect bilirubin increases |
| Increased bilirubin metabolism in the intestines | Stool is usually not discolored |
| Long-term chronic hemolysis | The risk of pigment gallstones increases |
| Severe intravascular hemolysis | Hemoglobinuria and kidney damage are possible. |
Source of the table. [73] [74] [75]
Symptoms
The clinical picture consists of two groups of symptoms: those of jaundice itself and those of hemolytic anemia. The patient may notice yellowing of the sclera and skin, but simultaneously complain of weakness, fatigue, palpitations, dizziness, and decreased exercise tolerance. Merck clearly states that the systemic manifestations of hemolytic anemia are similar to those of other anemias and include weakness, pallor, and fatigue, while jaundice and scleral icterus are added as a consequence of hyperbilirubinemia. [76]
Hemolytic jaundice is more typically associated with a combination of yellowing of the sclera and anemic syndrome than with severe itching or completely light-colored stools. Itching and acholic stools are much more strongly associated with cholestasis and mechanical jaundice. Therefore, based on the combination of symptoms, one can assume whether the mechanism of hyperbilirubinemia is prehepatic, hepatic, or posthepatic. [77]
In acute hemolytic crises, symptoms progress rapidly. Chills, fever, back and abdominal pain, dark urine, hemoglobinuria, weakness to the point of fainting, and sometimes acute renal dysfunction are possible. This is especially pronounced in cases of glucose-6-phosphate dehydrogenase deficiency following a trigger or in cases of paroxysmal nocturnal hemoglobinuria. [78] [79] [80]
In some patients, the picture is chronic and milder over a long period of time. In hereditary spherocytosis or chronic autoimmune variants, periodic jaundice, splenomegaly, weakness, and repeated episodes of cholelithiasis may predominate. In such cases, jaundice appears not as a sudden acute event, but as a wave-like symptom of chronic hemolysis. [81] [82]
If the cause is sickle cell disease or malaria, jaundice is often accompanied by signs of the underlying disease: pain crises, infections, organ damage, fever. This once again demonstrates that the clinical appearance of hemolytic jaundice is determined not only by bilirubin, but also by the type of hemolysis. [83] [84]
| Symptom | What does it mean more often? |
|---|---|
| Yellow sclera and skin | Hyperbilirubinemia due to hemolysis |
| Weakness and fatigue | Anemic syndrome |
| Dark urine | Hemoglobinuria or increased urobilinogen, more often with intravascular hemolysis |
| Enlarged spleen | Chronic extravascular hemolysis |
| Back and abdominal pain | Possible hemolytic crisis |
| Attacks of biliary colic | Possible pigment stones in chronic hemolysis |
Source of the table. [85] [86] [87]
Classification, forms and stages
The most important practical classification divides hemolytic jaundice by the mechanism of hemolysis into extravascular and intravascular. In the extravascular variant, cell destruction occurs primarily in the spleen and liver. In the intravascular variant, it occurs directly within the vascular bed. This distinction affects urine color, the risk of hemoglobinuria, the likelihood of kidney damage, and the set of laboratory signs. [88] [89]
The second important classification is congenital and acquired forms. Congenital forms include membranopathy, enzymopathies, and hemoglobinopathies. Acquired forms include immune, infectious, toxic, mechanical, microangiopathic, and clonal variants. For the clinician, this is a very convenient scheme because it immediately suggests the next steps. [90]
Hemolysis can progress to acute, chronic, or episodic stages. The acute stage is characterized by a rapid rise in bilirubin, a drop in hemoglobin, and more dramatic clinical manifestations. The chronic stage often presents with intermittent jaundice, splenomegaly, and the gradual development of biliary complications. The episodic nature is particularly typical of glucose-6-phosphate dehydrogenase deficiency, where the crisis is triggered by a trigger. [91] [92]
There is no formal, generally accepted staging system for hemolytic jaundice as a syndrome. In practice, severity is assessed based on the severity of anemia, the rate of hemolysis, bilirubin levels, reticulocytosis, involvement of the kidneys, spleen, and other organs, as well as whether the condition is compensated or a crisis. This is the working clinical analogue of staging. [93]
| Classification principle | Options |
|---|---|
| By the site of hemolysis | Extravascular, intravascular |
| By origin | Congenital, acquired |
| With the flow | Acute, chronic, episodic |
| By clinical severity | Compensated hemolysis, hemolytic crisis, organ complications |
Source of the table. [94] [95]
Complications and consequences
One of the most common complications of chronic hemolytic jaundice is pigment cholelithiasis. NIDDK and Merck indicate that black pigment stones consist of bilirubin compounds and develop more often in patients with hemolytic anemia. Clinically, this manifests as biliary colic, cholecystitis, and sometimes choledocholithiasis. [96] [97]
The second major problem is progressive anemia with tissue hypoxia. If the bone marrow fails to compensate for the destruction of red blood cells, decompensated hemolytic anemia develops. In severe cases, this leads to severe weakness, shortness of breath, tachycardia, fainting, and the risk of cardiac decompensation. [98]
Intravascular hemolysis increases the risk of kidney damage. This is particularly noticeable in glucose-6-phosphate dehydrogenase deficiency with massive crisis, paroxysmal nocturnal hemoglobinuria, and some microangiopathies. Hemoglobinuria and free hemoglobin can increase tubular damage and lead to acute kidney injury. [99] [100]
Some forms of hemolytic anemia are also characterized by a high thrombotic risk. This applies, for example, to autoimmune hemolytic anemia and paroxysmal nocturnal hemoglobinuria. In these cases, jaundice may only be a visible part of the process, while the main prognostic risk is associated with venous and arterial thrombosis. [101] [102]
| Complication | In what forms is it especially important? |
|---|---|
| Pigment gallstones | Chronic hemolytic anemia |
| Decompensated anemia | Any severe or rapid hemolysis |
| Acute kidney injury | Intravascular hemolysis, hemoglobinuria |
| Thrombosis | Autoimmune hemolytic anemia, paroxysmal nocturnal hemoglobinuria |
| Splenomegaly and hypersplenism | Extravascular chronic forms |
Source of the table. [103] [104] [105] [106]
When to see a doctor
A doctor should be consulted if any new yellowing of the sclera or skin occurs, especially if it is accompanied by weakness, pallor, dark urine, an enlarged spleen, or abdominal or back pain. Even if the jaundice appears moderate, it is impossible to determine whether it is benign hyperbilirubinemia or rapid hemolysis with the risk of complications without a blood test. [107] [108]
Urgent care is needed if jaundice develops rapidly and is accompanied by a sharp decline in health, shortness of breath, tachycardia, chest pain, severely dark urine, fainting, high fever, or decreased urine output. These signs may be consistent with a hemolytic crisis, acute kidney injury, thrombotic microangiopathy, severe infection, or massive immune hemolysis. [109] [110] [111]
A particularly rapid response is required in pregnant women, children, the elderly, patients with known hemolytic diseases, and people starting new medications. These groups are at higher risk for severe disease progression and diagnostic errors, and the window of safe observation is shorter. [112] [113]
Diagnostics
Diagnosis begins with confirming hemolysis itself. Merck recommends evaluating a complete blood count, reticulocytes, peripheral blood smear, indirect bilirubin, lactate dehydrogenase, haptoglobin, and urinalysis. If the picture indicates accelerated red blood cell destruction, the next step is to search for the cause. [114]
Bilirubin fractions help differentiate hemolytic jaundice from hepatic and mechanical jaundice. Hemolysis is typically characterized by a predominance of indirect bilirubin. The Merck Guide to Jaundice in Adults explicitly states that unconjugated hyperbilirubinemia is more often associated with increased bilirubin formation, decreased hepatic uptake, or impaired conjugation. In the context of anemia and hemolysis, the first of these mechanisms is most often the key one. [115]
A peripheral blood smear has a very high diagnostic value. Spherocytes suggest hereditary spherocytosis or warm autoimmune hemolytic anemia. Schistocytes indicate microangiopathic or mechanical hemolysis. Sickle-shaped red blood cells indicate sickle cell disease. The presence of agglutination may indicate the cold autoimmune form. [116] [117] [118]
The direct antiglobulin test is particularly important for confirming the immune origin of hemolysis. Merck emphasizes that it is this test that establishes the diagnosis of autoimmune hemolytic anemia and can suggest its type. If the test is negative and hemolysis is present, the physician shifts the search to membranopathies, enzymopathies, hemoglobinopathies, mechanical hemolysis, and paroxysmal nocturnal hemoglobinuria. [119]
Further steps depend on the suspected cause. For glucose-6-phosphate dehydrogenase deficiency, an enzyme analysis is needed. For paroxysmal nocturnal hemoglobinuria, flow cytometry is used. If hemoglobinopathy is suspected, electrophoresis or molecular diagnostics are used. For chronic biliary complications and pain in the right hypochondrium, an ultrasound examination of the gallbladder and bile ducts is included to detect pigment stones. [120] [121] [122]
| Diagnostic stage | What does it give? |
|---|---|
| Complete blood count and reticulocytes | Confirm anemia and regenerative response |
| Indirect bilirubin, lactate dehydrogenase, haptoglobin | Confirm hemolysis |
| Peripheral smear | Suggests the type of hemolytic anemia |
| Direct antiglobulin test | Confirms immune hemolysis |
| Urine analysis | Shows urobilinogen, hemoglobinuria |
| Special tests | Specify enzyme deficiencies, hemoglobinopathies, paroxysmal nocturnal hemoglobinuria |
| Ultrasound examination of the gallbladder | Detects pigment stones in chronic hemolysis |
Source of the table. [123] [124] [125] [126]
Differential diagnosis
The primary goal of differential diagnosis is to distinguish hemolytic jaundice from hepatic and mechanical jaundice. In the hemolytic form, indirect bilirubin usually predominates, while pronounced cholestatic signs, such as intense itching and light-colored stools, are less common. In mechanical jaundice, on the other hand, impaired bile flow is the primary concern, while in hepatocellular jaundice, damage to the liver tissue itself is the primary concern. [127]
The second important question is whether the patient actually has hemolysis, rather than isolated indirect hyperbilirubinemia without hemolysis, such as Gilbert's syndrome. Reticulocytes, haptoglobin, lactate dehydrogenase, a blood smear, and a complete blood count are helpful here. If there is no anemia, the smear is normal, and hemolysis is not confirmed by laboratory tests, the physician should consider more than just hemolytic jaundice. [128] [129]
The third line of differentiation lies within the hemolytic anemias themselves. It is necessary to distinguish between immune and non-immune forms, hereditary and acquired, and intravascular and extravascular hemolysis. A direct antiglobulin test, smear, family history, drug history, information on prosthetic valves, and the results of specialized tests are critical here. [130] [131]
The physician must also be aware of conditions that mimic severe hemolysis. For example, in some microangiopathies, schistocytes and jaundice may be associated with severe kidney damage and thrombocytopenia, while in severe infections such as malaria, jaundice may be part of a complex mixed picture with hemolysis and liver stress simultaneously. Therefore, a definitive diagnosis is almost always based on a combination of tests, rather than a single symptom. [132] [133]
| What needs to be distinguished | What helps? |
|---|---|
| Hemolytic and mechanical jaundice | Bilirubin fractions, stool color, signs of cholestasis |
| Hemolytic and hepatic jaundice | Liver function tests, hemolysis markers, clinical features |
| Hemolysis and Gilbert's syndrome | Presence of anemia, reticulocytosis, drop in haptoglobin |
| Immune and nonimmune hemolysis | Direct antiglobulin test |
| Hereditary and acquired hemolysis | Family history, smear, enzyme and genetic tests |
Source of the table. [134] [135] [136] [137]
Treatment
Treatment of hemolytic jaundice always begins with an assessment of the severity of hemolysis and the patient's general condition. If there are signs of a hemolytic crisis, such as severe anemia, hemoglobinuria, renal damage, thrombosis, high fever, or a rapid drop in hemoglobin, immediate treatment is required. Jaundice itself is usually not treated separately, as it improves when red blood cell destruction is slowed or stopped. [138] [139]
Basic support includes eliminating triggers, adequate hydration, monitoring renal function, folic acid for chronic hemolysis, and red blood cell transfusions when indicated. Merck emphasizes that hemolytic anemia with unexpectedly low reticulocytosis can become a hematological emergency and require urgent transfusion. This is especially important in severely ill patients whose bone marrow is temporarily unable to compensate. [140]
In warm autoimmune hemolytic anemia, glucocorticoids remain the first-line therapy. Merck and hematology guidelines indicate that they are traditionally used first, with rituximab, immunosuppressants, and, in some cases, splenectomy being used in insufficient response. Rituximab has gained a significant place in modern practice as an effective second-line option and, sometimes, for earlier intensification of therapy. [141] [142]
In cold agglutinin disease, cold abstinence alone is not enough. In addition to avoiding hypothermia and treating triggers, modern therapy includes targeted complement suppression. The US Food and Drug Administration (FDA) has approved sutimlimab for the treatment of hemolysis in adults with cold agglutinin disease. This is an important example of how treatment for hemolytic jaundice has become more targeted. [143] [144]
In hereditary spherocytosis, treatment depends on its severity. Mild forms sometimes require only observation and folate supplementation. In moderate and severe cases, splenectomy is considered, especially if anemia is severe, crises are frequent, or biliary complications have developed. However, the decision to perform splenectomy is always a balanced one, as reducing hemolysis comes at the cost of altered immune defenses and the need for vaccinations. [145] [146]
In glucose-6-phosphate dehydrogenase deficiency, the main principle is to eliminate the trigger as quickly as possible. This could be an infection, a drug, or other oxidative stress. Most episodes are reversible with adequate support, but severe hemolysis may require transfusion and renal monitoring, as Merck notes the risk of hemoglobinuria and acute kidney injury in more severe cases. [147]
In sickle cell disease, jaundice is often part of chronic hemolysis, and treatment is focused on controlling the disease itself. The World Health Organization recommends hydroxyurea, blood transfusions, infection prophylaxis, and, in some cases, bone marrow transplantation and gene therapy. Jaundice is reduced when the frequency of hemolytic and vaso-occlusive episodes is reduced. [148] [149]
For paroxysmal nocturnal hemoglobinuria, current therapy relies on complement inhibitors. Merck reports that eculizumab, ravulizumab, pegcetacoplan, and iptacopan reduce hemolysis, lower transfusion requirements, and lower thrombotic risk. For patients with this disease, jaundice is only part of the systemic complement-mediated pathology, so targeted therapy radically changes the prognosis. [150]
If jaundice develops due to microangiopathic hemolysis, such as thrombotic thrombocytopenic purpura (TTP), the approach is entirely different. In this situation, urgent hematological care becomes paramount, including plasma exchange, glucocorticoids, and specialized regimens such as caplacizumab for confirmed or highly probable TTP. Here, delay is more dangerous than the jaundice itself. [151] [152]
In chronic hemolysis, secondary complications, especially gallstones, often need to be treated. If biliary colic, cholecystitis, or choledocholithiasis develops in the presence of pigment stones, the treatment strategy is supplemented by gastroenterological or surgical interventions. This is important, because in some patients, it is the biliary complication, not the hemolysis itself, that leads to hospitalization. [153] [154]
| Clinical situation | The basic approach |
|---|---|
| Mild chronic hemolysis | Monitoring, folates, trigger control |
| Warm autoimmune hemolytic anemia | Glucocorticoids, then rituximab and other lines |
| Cold agglutinin disease | Avoidance of cold, treatment of triggers, and sutimlimab as indicated |
| Hereditary spherocytosis | Support, in severe cases splenectomy |
| Glucose-6-phosphate dehydrogenase deficiency | Eliminate the trigger, support, sometimes transfusion |
| Sickle cell disease | Hydroxyurea, transfusions, prevention of complications |
| Paroxysmal nocturnal hemoglobinuria | Complement inhibitors |
| Thrombotic microangiopathy | Urgent specialized hematological therapy |
| Biliary complications of chronic hemolysis | Treatment of gallstones and their consequences |
Source of the table. [155] [156] [ 157 ] [158 ] [159] [160]
Prevention
Prevention of hemolytic jaundice depends on the cause. For hereditary diseases, hemolysis itself cannot be completely prevented, but it is possible to reduce the frequency of crises, identify complications early, and prevent biliary and infectious complications. For patients with hereditary spherocytosis and sickle cell disease, this means regular monitoring, vaccinations as indicated, and monitoring for anemia and cholelithiasis. [161] [162]
For glucose-6-phosphate dehydrogenase deficiency, prevention is particularly practical. It revolves around strict avoidance of triggers: potentially dangerous medications, oxidative stress, and, in the classic case, fava beans. Merck and MedlinePlus emphasize that knowledge of triggers is crucial in these patients. [163] [164]
In autoimmune hemolytic anemia, relapse prevention involves controlling the underlying autoimmune or lymphoproliferative process and caution with medications that have previously caused immune hemolysis. For cold agglutinin disease, daily protection from cold is also important. Prevention here does not eliminate the need for treatment, but it significantly reduces the clinical burden. [165]
Early detection of biliary complications of chronic hemolysis is of particular preventative importance. If a patient experiences attacks of pain in the right hypochondrium, nausea after eating, or signs of cholestasis due to long-standing hemolytic disease, an examination should not be delayed. This is how more severe complications, including cholecystitis and choledocholithiasis, can be prevented. [166] [167]
| Prevention direction | For whom is it especially important? |
|---|---|
| Avoiding oxidative triggers | Glucose-6-phosphate dehydrogenase deficiency |
| Control of chronic hemolytic disease | Hereditary spherocytosis, sickle cell disease |
| Infection prevention and vaccination | After splenectomy and in a number of chronic forms |
| Avoiding the cold | Cold agglutinin disease |
| Early control of biliary symptoms | Any long-term chronic hemolysis |
Source of the table. [168] [169] [170] [171]
Forecast
The prognosis is determined not by the bilirubin level itself, but by the underlying cause of hemolysis, the speed of diagnosis, and the availability of specific treatment. In reversible triggered forms, such as an episode of hemolysis in a patient with glucose-6-phosphate dehydrogenase deficiency, the outcome is often good if the triggering factor is quickly removed and the kidneys and hemodynamics are supported. [172]
In chronic hereditary diseases, the prognosis is more variable. Many patients with hereditary spherocytosis live long lives, but may suffer from recurrent jaundice, anemia, and gallstones. In sickle cell disease, the prognosis depends not only on hemolysis but also on vascular, infectious, and organ complications, although modern treatments have significantly improved outcomes. [173] [174] [175]
For autoimmune hemolytic anemia, the prognosis depends on the disease's form, response to therapy, and the presence of a secondary cause. For paroxysmal nocturnal hemoglobinuria, the advent of complement inhibitors has radically altered the prognosis, reducing the severity of hemolysis and thrombotic risk. Thus, for a number of causes of hemolytic jaundice, modern targeted therapy has indeed altered the natural course of the disease. [176] [177]
A poor prognosis is often associated with late recognition of severe intravascular hemolysis, microangiopathy, infections, and thrombosis. Therefore, the most important prognostic factor is not "how yellow the patient is," but how quickly an accurate diagnosis is made and causal treatment is initiated. [178] [179]
| Forecast factor | How does it affect |
|---|---|
| Quick identification of the cause | Improves outcome |
| Trigger reversibility | Makes hemolysis more controllable |
| Chronic hemolysis | Increases the risk of biliary complications |
| Availability of targeted therapy | Significantly improves the prognosis for some diseases |
| Late diagnosis of microangiopathy or severe intravascular hemolysis | Worsens the outcome |
Source of the table. [180] [181] [182]
FAQ
Can hemolytic jaundice occur without severe pain?
Yes. In chronic extravascular hemolysis, jaundice can be moderate and almost painless, especially if anemia and splenomegaly predominate. Pain is more common with hemolytic crises, malaria, hemoglobinuria, or cholelithiasis. [183] [184]
Is dark urine always associated with hemolytic jaundice?
Not always. Dark urine is more characteristic of intravascular hemolysis with hemoglobinuria or a marked increase in urobilinogen. With predominantly extravascular hemolysis, this symptom may be less noticeable. [185] [186]
Is it possible to differentiate hemolytic jaundice from mechanical jaundice by appearance alone?
No. There are clinical clues, but they can only be reliably distinguished by testing and, if necessary, imaging. For the hemolytic form, signs of hemolysis and indirect bilirubin are more important, while for mechanical jaundice, cholestatic changes and signs of bile duct obstruction are more important. [187] [188]
Why do gallstones form with chronic hemolysis?
Because more bilirubin pigments enter the bile, and over time, black pigment stones form. This is a classic complication of long-term hemolytic anemia. [189] [190]
Is it necessary to treat bilirubin itself if the cause of hemolysis has already been identified?
Typically, the primary focus is on controlling hemolysis. When red blood cell destruction decreases, bilirubin levels also decrease. Reducing bilirubin levels alone without addressing the underlying cause rarely solves the problem. [191]
Is hemolytic jaundice life-threatening?
Yes, especially in cases of severe autoimmune hemolysis, thrombotic microangiopathies, paroxysmal nocturnal hemoglobinuria, massive hemoglobinuria, and infections such as severe malaria. The danger is determined not only by the jaundice itself, but also by the rate of red blood cell destruction and organ damage. [192] [193] [194]
Key points from experts
Gloria F. Gerber, MD, a hematologist at Johns Hopkins School of Medicine, is the author and editor of the Merck Manual's professional sections on hemolytic anemias. Her materials consistently demonstrate a central clinical principle: hemolytic jaundice is a consequence of accelerated red blood cell destruction, and the diagnostic center of gravity lies in confirming hemolysis, its mechanism, and rate, not just in the bilirubin level. [195] [196] [197]
Marie Scully, Professor of Haematology at University College London Hospitals, National Service Director for Thrombotic Thrombocytopenic Purpura and Associated Thrombotic Microangiopathies, and lead author of the UK guidelines, is particularly relevant to this topic: in cases of jaundice secondary to haemolysis, microangiopathies should always be considered as a potential emergency, where delay is more dangerous than hyperbilirubinemia itself. [198]
Bhaskar K. Somani, Professor of Urology at University Hospital Southampton, is a European Association of Urology expert on urolithiasis. Although he does not focus on hemolytic anemia as a primary topic, the urological portion of his work highlights another important practical implication: chronic hemolysis must be assessed not only from a hematological perspective but also from the perspective of biliary complications, as pigment stones and their sequelae are significantly more common in such patients. [199]
Conclusion
Hemolytic jaundice is a clinical sign that red blood cells are being destroyed faster than the liver can process bilirubin. It is a prehepatic form of jaundice and is most often accompanied by elevated indirect bilirubin, anemia, reticulocytosis, and signs of active hemolysis. However, this general mechanism conceals a variety of diseases—from relatively stable hereditary forms to life-threatening immune and microangiopathic conditions. [200] [201]
The safest and most modern approach is to look beyond the word "jaundice." It's important to quickly determine whether hemolysis is present, whether it's intravascular or extravascular, congenital or acquired, immune or non-immune, and whether there are any renal, thrombotic, or biliary complications. Accurate diagnosis of the underlying cause is what truly makes treatment effective and changes the prognosis. [202] [203] [204]