Medical expert of the article

New publications
Leah's subacute necrotizing encephalomyopathy
Last reviewed: 04.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
 [
[ Causes of Leah's syndrome
The disease is based on a deficiency of enzymes that provide energy production mainly due to a disruption of pyruvic acid metabolism and a defect in electron transport in the respiratory chain. A deficiency of the pyruvate dehydrogenase complex (a-E1 subunit), pyruvate carboxylase, complex 1 (NAD-coenzyme Q-reductase) and complex 4 (cytochrome oxidase) of the respiratory chain develops.
It has been established that defects of pyruvate carboxylase, complex 1 (NAD-coenzyme Q-reductase) and complex 4 (cytochrome oxidase) of the respiratory chain are inherited in an autosomal recessive manner, defects of the pyruvate dehydrogenase complex (a-E1 subunit) are inherited in an X-linked recessive manner. In case of point mutations of mtDNA, which affect the 6th subunit of ATPase, mitochondrial inheritance is typical. Most often, a miscens mutation occurs, associated with the replacement of thymine with guanine or cytosine at position 8993 of mtDNA. Less common is a mutation at position 9176 of mtDNA. Due to the fact that the T8993G mutation is the main defect in NARP syndrome, families with these two diseases have been described. In children, a mutation in mtDNA at position 8344 has also been described, which occurs in MERRF syndrome.
It is assumed that in the case of accumulation of mutant mtDNA in most mitochondria, a severe course of Leigh syndrome develops. In the mitochondrial genesis of this condition, mutant mtDNA is found in 90% of all mitochondria. The pathogenesis is associated with a violation of energy formation in cells and the development of lactic acidosis.
Symptoms of Leah's syndrome
The first signs of the disease debut at an early age (1-3 years). However, there are known cases of the disease manifestation at 2 weeks and at 6-7 years of age. At first, nonspecific disorders develop: delayed psychomotor development, decreased appetite, episodes of vomiting, body weight deficit. Subsequently, neurological symptoms increase: muscle hypotonia or dystonia with transition to hypertonia, attacks of myoclonus or tonic-clonic seizures, tremor of the extremities, choreoathetosis, coordination disorder, decreased tendon reflexes, lethargy, drowsiness. Cerebral neurodegeneration is progressive. Symptoms of pyramidal and extrapyramidal insufficiency increase, the act of swallowing is impaired. Such changes in the organ of vision as ptosis, ophthalmoplegia, atrophy of the optic nerves, less often pigmentary degeneration of the retina are often observed. Sometimes hypertrophic cardiomyopathy develops, episodes of tachypnea appear.
Rarely, the disease proceeds as an acute encephalopathy. More typical is a chronic or subacute course, which leads to a fatal outcome several years after the onset of the disease. With a rapid course (several weeks), death occurs as a result of paralysis of the respiratory center.
Diagnostics of Leah's syndrome
A biochemical blood test reveals lactic acidosis due to the accumulation of lactic and pyruvic acids in the blood and cerebrospinal fluid, as well as an increase in the alanine content in the blood. The level of ketone bodies may also be elevated. Increased excretion of organic acids is detected in the urine: lactic, fumaric, etc. The level of carnitine in the blood and tissues often decreases.
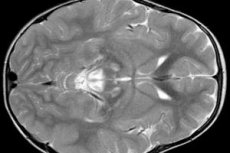
EEG results reveal focal signs of epileptic activity. MRI data reveals enlargement of the cerebral ventricles, bilateral brain damage, calcification of the basal ganglia (caudate nucleus, putamen, substantia nigra, globus pallidus). Atrophy of the cerebral hemispheres and brain matter can also be detected.
Morphological examination reveals gross changes in the brain matter: symmetrical foci of necrosis, demyelination and spongy degeneration of the brain, mainly of the middle sections, pons, basal ganglia, thalamus and optic nerve. The histological picture includes cystic degeneration of brain tissue, astrocytic gliosis, neuronal death and an increase in the number of mitochondria in cells. In skeletal muscles, there is accumulation of lipid inclusions, a decrease in the histochemical reaction to complexes 1 and 4 of the respiratory chain, subsarcolemmal accumulation of mitochondria, abnormal mitochondria with disorganization of the cristae. The RRF phenomenon is often not detected.
How to examine?
What tests are needed?
Использованная литература