Medical expert of the article

New publications
Polymyositis and dermatomyositis: causes, symptoms, diagnosis, treatment
Last reviewed: 05.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
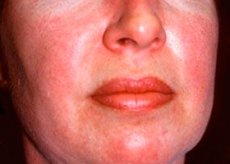
Polymyositis and dermatomyositis are rare systemic rheumatic diseases characterized by inflammatory and degenerative changes in muscles (polymyositis) or muscles and skin (dermatomyositis). The most specific skin manifestation is heliotrope rash.
Muscle involvement is symmetrical and includes weakness, some tenderness, and subsequent atrophy of the proximal pelvic girdle muscles. Complications may include visceral involvement and malignancy. Diagnosis is based on clinical presentation and assessment of muscle dysfunction by measuring enzyme levels, performing MRI, electromyography, and muscle biopsy. Treatment involves glucocorticoids, sometimes in combination with immunosuppressants or intravenous immunoglobulins.
Women get sick twice as often as men. The disease can occur at any age, but is most often detected in the range from 40 to 60 years; in children - from 5 to 15 years.
 [
[ What causes dermatomyositis and polymyositis?
The cause of the disease is believed to be an autoimmune reaction to muscle tissue in genetically predisposed individuals. The disease is more common in the presence of a burdened family history and carriers of certain HLA antigens (DR3, DR52, DR56). Possible triggers are viral myositis and malignant neoplasms. There are reports of detection of structures similar to picornaviruses in muscle cells; in addition, viruses can induce similar diseases in animals. The association of malignant tumors with dermatomyositis (much less frequently than with polymyositis) suggests that tumor growth may also be a trigger for the development of the disease as a result of the initiation of autoimmune reactions to common antigens of the tumor and muscle tissue.
Deposits of IgM, IgG, and the third component of complement are found in the walls of blood vessels of skeletal muscles; this is especially characteristic of dermatomyositis in children. Patients with polymyositis may also develop other autoimmune processes.
Pathophysiology of dermatomyositis and polymyositis
Pathological changes include cell damage and atrophy against the background of inflammation of varying degrees of severity. The muscles of the upper and lower extremities, as well as the face, are damaged to a lesser extent than other skeletal muscles. Damage to the visceral muscles of the pharynx and upper esophagus, less often the heart, stomach or intestines, can lead to dysfunction of the above organs. High concentrations of myoglobin due to rhabdomyolysis can cause kidney damage. Inflammatory changes in the joints and lungs may also occur, especially in patients who have antisynthetase antibodies.
Symptoms of dermatomyositis and polymyositis
The onset of polymyositis may be acute (especially in children) or subacute (more often in adults). Acute viral infection sometimes precedes or is a trigger for the manifestation of the disease, the most common manifestations of which are weakness of the proximal muscles or skin rashes. Pain is expressed to a lesser extent than weakness. Polyarthralgia, Raynaud's phenomenon, dysphagia, lung disorders, general symptoms (increased body temperature, decreased body weight, weakness) may develop. Raynaud's phenomenon is often found in patients with concomitant connective tissue diseases.
Muscle weakness may progress over several weeks or months. However, for clinical manifestation of muscle weakness, at least 50% of muscle fibers must be affected (thus, the presence of muscle weakness indicates the progression of myositis). Patients may experience difficulty raising their arms above shoulder level, walking up stairs, or rising from a sitting position. Due to severe weakness of the pelvic and shoulder girdle muscles, patients may be confined to a wheelchair or bed. If the flexors of the neck are affected, it becomes impossible to lift the head from the pillow. Affection of the muscles of the pharynx and upper esophagus leads to swallowing disorders and regurgitation. The muscles of the lower, upper limbs, and face are usually not affected. However, contractures of the limbs may develop.
Skin rashes observed in dermatomyositis are usually dark in color and erythematous. Periorbital edema of a purple color (heliotrope rash) is also characteristic. Skin rashes may be slightly elevated above the skin level and be smooth or scaly; localization of rashes is the forehead, neck, shoulders, chest, back, forearms, lower parts of the shins, eyebrows, knee areas, medial malleoli, dorsal surfaces of the interphalangeal and metacarpophalangeal joints, on the lateral side (Gottron's symptom). Hyperemia of the base or periphery of the nails is possible. Desquamative dermatitis, accompanied by cracks, may develop on the skin of the lateral surface of the fingers. Primary skin lesions often resolve without sequelae, but may lead to secondary changes such as dark pigmentation, atrophy, scarring, or vitiligo. Subcutaneous calcifications may develop, especially in children.
Approximately 30% of patients develop polyarthralgia or polyarthritis, often accompanied by edema and joint effusion. However, the severity of joint manifestations is small. They occur more often when antibodies to Jo-1 or other synthetases are detected in patients.
Involvement of internal organs (except the pharynx and upper esophagus) is less common in polymyositis than in other rheumatic diseases (particularly SLE and systemic sclerosis). Rarely, especially in antisynthetase syndrome, the disease manifests as interstitial pneumonitis (in the form of dyspnea and cough). Cardiac arrhythmias and conduction disturbances may develop, but they are usually asymptomatic. Gastrointestinal manifestations are more common in children who also have vasculitis and may include vomiting with blood, melena, and intestinal perforation.
Where does it hurt?
Classification of polymyositis
There are 5 types of polymyositis.
- Primary idiopathic polymyositis, which can occur at any age, does not involve the skin.
- Primary idiopathic dermatomyositis is similar to primary idiopathic polymyositis but involves the skin.
- Polymyositis and dermatomyositis associated with malignant neoplasms can occur in patients of any age; their development is most often observed in elderly patients, as well as in patients with other connective tissue diseases. The development of malignant neoplasms can be observed both within 2 years before and within 2 years after the onset of myositis.
- Childhood polymyositis or dermatomyositis is associated with systemic vasculitis.
- Polymyositis and dermatomyositis can also occur in patients with other connective tissue diseases, most commonly progressive systemic sclerosis, mixed connective tissue disease, and SLE.
The inclusion of myositis of the truncal muscles in the group of polymyositis is incorrect, since the latter is a separate disease characterized by clinical manifestations similar to those of chronic idiopathic polymyositis. However, it develops in old age, often affects the muscles of the distal parts of the body (for example, the upper and lower extremities), has a longer duration, responds less well to treatment, and is characterized by a typical histological picture.
Diagnosis of dermatomyositis and polymyositis
Polymyositis should be suspected in patients with complaints of proximal muscle weakness, with or without tenderness. Evaluation for dermatomyositis is necessary in patients with complaints of a rash resembling heliotrope or Gottron's sign, as well as in patients with manifestations of polymyositis combined with any skin lesions consistent with dermatomyositis. The clinical manifestations of polymyositis and dermatomyositis may resemble those of systemic sclerosis or, less commonly, SLE or vasculitis. The certainty of the diagnosis is increased by meeting as many of the following five criteria as possible:
- weakness of proximal muscles;
- characteristic skin rashes;
- increased activity of muscle tissue enzymes (creatine kinase or, in the absence of an increase in its activity, aminotransferases or aldolase);
- characteristic changes in myography or MRI;
- characteristic histological changes in a muscle tissue biopsy (absolute criterion).
Muscle biopsy can exclude some clinically similar conditions, such as myositis of the truncal muscles and rhabdomyolysis due to viral infection. Changes revealed by histological examination may vary, but chronic inflammation, foci of muscle degeneration and regeneration are typical. An accurate diagnosis (usually by histological verification) is necessary before starting potentially toxic treatment. MRI can detect foci of edema and inflammation in the muscles, followed by their targeted biopsy.
Laboratory studies can confirm or, on the contrary, eliminate the suspicion of the presence of the disease, and are also useful in assessing its severity, the possibility of a combination with other similar pathology and diagnosing complications. Despite the fact that antinuclear antibodies are detected in some patients, this phenomenon is more typical of other connective tissue diseases. About 60% of patients have antibodies to the antigen of nuclei (PM-1) or whole thymus cells and Jo-1. The role of autoantibodies in the pathogenesis of the disease remains unclear, although it is known that antibodies to Jo-1 are a specific marker of antisynthetase syndrome, including fibrosing alveolitis, pulmonary fibrosis, arthritis and Raynaud's phenomenon.
Periodic assessment of creatine kinase activity is useful for monitoring treatment. However, in patients with severe muscle wasting, the enzyme activity may be normal despite the presence of chronic active myositis. MRI, muscle biopsy, or elevated creatine kinase activity are often helpful in differentiating between relapse of polymyositis and glucocorticoid-induced myopathy.
Because many patients have undiagnosed malignancies, some authors recommend screening all adults with dermatomyositis and those with polymyositis over the age of 60 years using the following schedule: physical examination, including breast, pelvic, and rectal examinations (including stool testing for occult blood); complete blood count; blood chemistry; mammography; carcinoembryonic antigen testing; urinalysis; chest radiography. Some authors question the need for such screening in younger patients who do not have clinical evidence of malignancy.
What do need to examine?
How to examine?
Treatment of dermatomyositis and polymyositis
Until the inflammation is relieved, physical activity should be limited. Glucocorticoids are first-line drugs. In the acute stage of the disease, adult patients should be prescribed prednisolone (orally) at a dose of 40 to 60 mg per day. Regular determination of creatine kinase activity is an early indicator of effectiveness: in most patients, a decrease or normalization is noted within 6 to 12 weeks following an increase in muscle strength. After normalization of enzyme activity, the prednisolone dose is reduced: first by approximately 2.5 mg per day over a week, then more rapidly; if increased muscle enzyme activity relapses, the hormone dose is increased again. Recovered patients can do without glucocorticoids, but most often adult patients require long-term glucocorticoid therapy (10-15 mg of prednisolone per day). The initial dose of prednisolone for children is 30-60 mg/m 2 once a day. In the presence of remission for > 1 year in children, glucocorticoid therapy may be discontinued.
In some cases, patients receiving high doses of glucocorticoids experience a sudden increase in muscle weakness, which may be associated with the development of glucocorticoid myopathy.
In case of inadequate response to glucocorticoid treatment, as well as in case of development of glucocorticoid myopathy or other complications requiring dose reduction or discontinuation of prednisolone, immunosuppressants (methotrexate, cyclophosphamide, azathioprine, cyclosporine) should be used. Some patients may receive only methotrexate (usually in doses exceeding those used for RA treatment) for more than 5 years. Intravenous immunoglobulins may be effective in patients refractory to drug therapy, but their use increases the cost of treatment.
Myositis associated with primary and metastatic tumors, as well as myositis of the trunk muscles, are usually more refractory to glucocorticoid therapy. Remission of myositis associated with malignant tumors is possible after tumor removal.
What is the prognosis for dermatomyositis and polymyositis?
Long-term remission (and even clinical recovery) over 5 years is observed in more than half of treated patients; in children, this figure is higher. Relapse, however, can occur at any time. The overall five-year survival rate is 75%, higher in children. The causes of death in adults are severe and progressive muscle weakness, dysphagia, decreased nutrition, aspiration pneumonia or respiratory failure due to lung infections. Polymyositis is more severe and resistant to treatment if there is damage to the heart and lungs. Death in children can occur due to intestinal vasculitis. The general prognosis of the disease is also determined by the presence of malignant neoplasms.