Medical expert of the article

New publications
Mucopolysaccharidosis type 3
Last reviewed: 04.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
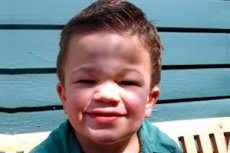
Mucopolysaccharidosis, type III (synonyms: Sanfilippo syndrome, lysosomal a-N-acetylglucosaminidase deficiency - mucopolysaccharidosis III A, acetyl-CoA-a-glucosaminid-N-acetyltransferase deficiency - mucopolysaccharidosis III B, N-acetylglucosamine-6-sulfatase - mucopolysaccharidosis III C, sulfamidase deficiency - mucopolysaccharidosis III D).
 [
[ Pathogenesis
The disease is caused by mutations in four different genes: lysosomal a-N-acetylglucosaminidase (mucopolysaccharidosis III A), acetyl-CoA-a-glucosaminid-N-acetyltransferase (mucopolysaccharidosis III B), lysosomal N-acetylglucosamine-6-sulfatase (mucopolysaccharidosis III C), and sulfamidase (mucopolysaccharidosis III D). All enzymes are involved in the metabolism of heparan sulfate.
The heparan-N-sulfatase gene - SGSH - is located on the long arm of chromosome 17 - 17q25.3. 75.3% of currently known mutations in the SGSH gene are point mutations. Frequently occurring mutations characteristic of European populations have been described - R74C (56% in Poland and 21% in Germany) and R245H (56% in the Netherlands).
The frequency of the R74C mutation is 47.5%, the R245H mutation is 7.5%. The other two described mutations, delll35G and N389S, together account for 21.7% of mutant alleles.
The gene for aN-acetyl-glucosaminidase (NAGLU) is located on the long arm of chromosome 17 - 17q21. 69% of mutations found in the NAGLU gene are missense and nonsense mutations, 26.3% are small deletions and insertions. The gene for acetyl-CoA-cc-glucosaminid-N-acetyltransferase (HGSNAT) is located on the short arm of chromosome 8 - 8p11.1. The gene was characterized only in 2006 and to date only a few mutations have been found in it.
The N-acetyl-glucosamine-6-sulfatase gene - GNS - is located on the long arm of chromosome 12 - 12ql4. There are 12 patients with mucopolysaccharidosis IIID registered in the world. Four mutations in the GNS gene have been described.
In all subtypes of mucopolysaccharidosis III, there is a disturbance in the degradation of heparan sulfate, which is part of the structure of cell membranes, including neuronal membranes, which correlates with a severe neurodegenerative process caused by cortical atrophy. Chronic diarrhea is explained by the involvement of the autonomic nervous system in the pathological process along with dysfunction of the intestinal mucosa. Sensorineural hearing loss is probably due to three causes: frequent otitis, deformation of the auditory ossicles and anomalies of the inner ear. Joint stiffness is the result of deformation of the metaphyses, thickening of the joint capsule is secondary to the deposition of glycosaminoglycans and fibrosis. Intra-syndromic differences in the severity of the disease are due exclusively to the residual functional activity of the mutant enzyme: the higher it is, the milder the disease.
Symptoms mucopolysaccharidosis type 3.
Clinical polymorphism in Sanfilippo syndrome is less pronounced than in other types of mucopolysaccharidosis. Slow progression of the disease, severe neurological disorders with mild symptoms from internal organs and other systems are characteristic.
The first symptoms of the disease usually appear between the ages of 2 and 6 in children with previously normal development. Manifest symptoms include regression of psychomotor and speech development, psychiatric disorders in the form of hyperactivity syndrome, autistic or aggressive behavior, sleep disorders; children become careless and inattentive.
Other common symptoms are hirsutism, coarse hair, moderate hepatosplenomegaly, valgus deformity of the limbs, and a short neck. The development of coarse facial features such as gargoyilism and skeletal deformities such as multiple dysostosis are weakly expressed in mucopolysaccharidosis III compared to other types of mucopolysaccharidosis characterized by the Hurler phenotype. Height, as a rule, corresponds to age, and joint stiffness rarely causes dysfunction. Most patients often develop osteoporosis and osteomalacia. Secondary skeletal disorders - a high risk of pathological fractures. Severe psychoneurological disorders are most often observed by the 6th-10th year of life, they lead to pronounced social maladjustment. Progressive sensorineural hearing loss is inherent in all patients with severe and moderate forms of the disease. Convulsions are observed in almost all patients as the disease progresses.
The disease progresses rapidly, and most patients do not survive to age 20. Mucopolysaccharidosis IIIA is considered to be the most common and severe type of this syndrome.
Diagnostics mucopolysaccharidosis type 3.
The diagnosis of mucopolysaccharidosis III is confirmed by determining the level of urinary glycosaminoglycan excretion and measuring enzyme activity. In the case of mucopolysaccharidosis III, the total excretion of glycosaminoglycans in the urine increases and hyperexcretion of heparan sulfate is observed. The activity of lysosomal enzymes corresponding to a certain subtype of mucopolysaccharidosis III is measured in leukocytes or skin fibroblast culture using an artificial fluorogenic substrate.
Prenatal diagnostics is possible by measuring enzyme activity in chorionic villus biopsy at 9-11 weeks of pregnancy and/or determining the spectrum of glycosaminoglycans in amniotic fluid at 20-22 weeks of pregnancy. For families with a known genotype, DNA diagnostics can be performed in early pregnancy.
What tests are needed?
Differential diagnosis
Differential diagnostics are carried out both within the group of mucopolysaccharidoses and with other lysosomal storage diseases: mucolipidoses, galactosialidosis, sialidosis, mannosidosis, fucosidosis, GM1 gangliosidosis.
Who to contact?
Treatment mucopolysaccharidosis type 3.
To date, effective methods of therapy for mucopolysaccharidosis III have not been developed. Symptomatic therapy is indicated.
Использованная литература