
Hereditary nephritis (Alport syndrome) in children
Last reviewed: 23.04.2024

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
Hereditary nephritis (Alport syndrome) is a genetically determined hereditary non-immune glomerulopathy that exhibits hematuria (sometimes with proteinuria), a progressive decrease in renal function with the development of chronic renal failure, often combined with neurosensory deafness and visual impairment.
For the first time the disease was described in 1902 by LGGuthrie, who observed a family in several generations of which hematuria was observed. In 1915, members of the same AFHurst family described the development of uremia. In 1927, A Alport first identified deafness in several relatives with hematuria. In the 50s of the last century, eye injuries were described in such a disease. In 1972, in patients with hereditary hematuria, morphologically examining renal tissue, Hinglais et al. Revealed uneven expansion and delamination of glomerular basal membranes. In 1985, the genetic basis of hereditary nephritis - a mutation in the gene of type IV collagen (Fiengold et al., 1985) was identified.
Investigation of the genetic nature of the disease made it possible to conclude that differences in phenotypic manifestations of hereditary nephritis (with or without hearing loss) are due to the degree of expression of the mutant gene. Thus, at present all clinical variants are considered as manifestations of one disease and the term "hereditary nephritis" is synonymous with the term "Alport syndrome".
According to epidemiological studies, hereditary nephritis occurs at a frequency of 17 per 100,000 children.
Causes of Alport syndrome
The genetic basis of the disease is a mutation in gene a-5 of the collagen chain of type IV. This type is universal for the basal membranes of the kidney, cochlear, lens capsule, retina and the cornea of the eye, which is proven in studies using monoclonal antibodies against this collagen fraction. Recently, they indicate the possibility of using DNA probes for prenatal diagnosis of hereditary nephritis.
The importance of testing all members of the family using DNA probes to identify carriers of the mutant gene is emphasized, which is of great importance in conducting medical genetic counseling of families with this disease. However, up to 20% of families do not have relatives with kidney disease, which suggests a high incidence of spontaneous mutations in the abnormal gene. The majority of patients with hereditary nephritis in families have individuals with kidney disease, hearing loss and vision pathology; related marriages between people who have one or more ancestors, since the marriage of related individuals increases the likelihood of obtaining the same genes from both parents. Autosomal dominant and autosomal recessive and dominant, linked to the X chromosome of the transmission pathway are established.
Children are more likely to distinguish three variants of hereditary nephritis: Alport syndrome, hereditary nephritis without hearing loss and family benign hematuria.
Alport Syndrome - hereditary nephritis with hearing damage. The basis is a combined defect in the structure of the collagen of the basal membrane of the glomeruli of the kidneys, the structures of the ear and eye. The gene of the classic Alport syndrome is located at the locus 21-22 q of the long arm of the X chromosome. In most cases, it is inherited by the dominant type linked to the X chromosome. In this regard, in men, Alport syndrome is more difficult, because in women the mutant gene function is compensated by a healthy allele of the second, intact chromosome.
Genetic basis of development of hereditary nephritis are mutations in the genes of alpha chains of type IV collagen. Six a-chains of IV type G collagen are known: the genes of a5- and a6-chains (Co4A5 and Co4A5) are located on the long arm of the X chromosome in the 21-22q zone; genes of a3- and a4-chains (Co4A3 and Co4A4) - on the 2-nd chromosome; genes of a1- and a2-chains (Co4A1 and Co4A2) - on the 13th chromosome.
In most cases (80-85%), an X-linked type of disease inheritance is associated with damage to the Co4A5 gene due to deletion, point mutations or splicing disorders. Currently, more than 200 mutations of the gene Kol4A5, responsible for the violation of synthesis of a5-chains of collagen type IV, are found. In this type of inheritance, the disease manifests itself in children of both sexes, but in boys it is more difficult.
Mutations in the loci of the genes Co4A3 and Co4A4, responsible for the synthesis of a3 and a4 - chains of type IV collagen, are inherited autosomally. According to research, autosomal dominant type of inheritance is observed in 16% of cases of hereditary nephritis, autosomal recessive - in 6% of patients. There are about 10 mutations of the genes of Co4A3 and Co4A4.
The result of mutations is a violation of the processes of assembly of type IV collagen, leading to a disruption in its structure. Collagen type IV is one of the main components of the glomerular basement membrane, the cochlear apparatus and the lens of the eye, the pathology of which will be revealed in the clinic of hereditary nephritis.
Collagen type IV, which is part of the glomerular basement membrane, consists mainly of two al-chains (IV) and one a2-chain (IV), and also contains a3, a4, a5-chains. Most often, with X-linked inheritance, the mutation of the Co4A5 gene is accompanied by the absence of a3, a4, a5 and a6 chains in the structure of type IV collagen, and the number of o1 and a2 chains in the glomerular basement membrane increases. The mechanism of this phenomenon is unclear, it is assumed that the cause is the posttranscriptional changes in mRNA.
The absence of a3-, a4- and a5-chains in the structure of type IV collagen of the basal glomeruli membranes leads to their thinning and brittleness in the early stages of Alport syndrome, which is clinically manifested more often by hematuria (less often hematuria with proteinuria or only proteinuria), hearing loss and lenticone. Further progression of the disease leads to a thickening and disruption of the permeability of the basal membranes in the late stages of the disease, with the proliferation of collagen V and VI types, manifested in the growth of proteinuria and the reduction of renal functions.
The nature of the mutation underlying hereditary nephritis largely determines its phenotypic manifestation. When deletion of the X chromosome with a simultaneous mutation of the genes Co4A5 and Co4A6, responsible for the synthesis of the a5 and a6 chains of type IV collagen, Alport syndrome is combined with the esophageal and genital organs leiomyomatosis. According to the research data, the mutation of the Co4A5 gene associated with deletion shows a great severity of the pathological process, a combination of renal damage with extrarenal manifestations and early development of chronic renal failure, compared to the stem mutation of this gene.
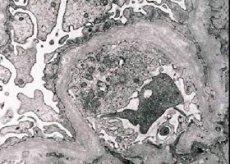
Morphologically, electron microscopy reveals the thinning and delamination of glomerular basal membranes (especially lamina densa) and the presence of electronically dense granules. The lesion of the glomerulus may be non-uniform in the same patient, from minimal focal lesion of mesangium to glomerulosclerosis. Glomerulitis in Alport syndrome is always immune-negative, which distinguishes it from glomerulonephritis. Characteristic are the development of canal atrophy, lymphohistiocyte infiltration, the presence of "foam cells" with inclusions of lipids - lipofagi. With the progression of the disease, a thickening and marked destruction of the basal glomeruli membranes are revealed.
Certain changes in the state of the immune system are revealed. In patients with hereditary nephritis, a decrease in the level of Ig A and a tendency to increase the concentration of IgM in the blood are noted, the level of IgG can be increased in the early stages of the development of the disease and decrease in late terms. Perhaps an increase in the concentration of IgM and G is a kind of compensatory response in response to a deficit of IgA.
The functional activity of the T-lymphocyte system is reduced; there is a selective decrease in B-lymphocytes responsible for the synthesis of Ig A, the phagocytic link of immunity is violated, mainly due to the violation of chemotaxis and intracellular digestion in neutrophils
In the study of the kidney biopsy in patients with Alport syndrome according to electron microscopy, ultrastructural changes in the basal glomerular membrane are observed: thinning, structural breakdown and splitting of glomerular basal membranes with a change in its thickness and uneven contours. In the early stages of hereditary nephritis, the defect determines the thinning and fragility of the glomerular basal membranes.
Thinning of glomerular membranes is a more favorable sign and is more common in girls. A more constant electron microscopic feature in hereditary nephritis is the cleavage of the basal membrane, and the severity of its destruction correlates with the severity of the process.

Symptoms of Alport Syndrome in Children
The first symptoms of Alport syndrome in the form of isolated urinary syndrome are more often detected in children of the first three years of life. In most cases, the disease is detected by accident. The urinary syndrome is revealed during the preventive examination of the child, before entering the children's institution or during ARVI. In case of appearance of pathology in the urine during ARVI. In hereditary nephritis, unlike the acquired glomerulonephritis, there is no latent period.
In the initial stage of the disease, the child's well-being suffers little, the characteristic feature is the persistence and persistence of the urinary syndrome. One of the main signs is hematuria of varying degrees, observed in 100% of cases. The increase in the degree of hematuria is noted during or after respiratory tract infections, physical exertion or after preventive vaccinations. Proteinuria in most cases does not exceed 1 g / day, at the onset of the disease may be unstable, as the process progresses proteinuria increases. Periodically, urinary sediment can have leukocyturia with a predominance of lymphocytes, which is associated with the development of interstitial changes.
Later, there is a violation of the partial functions of the kidneys, worsening of the general condition of the patient: intoxication, muscle weakness, arterial hypotension, often hearing impairment (especially in boys), sometimes impaired vision. Intoxication manifests as pallor, fatigue, headaches. In the initial stage of the disease, hearing loss in most cases is detected only by audiography. Hearing loss in Alport syndrome can occur in different periods of childhood, but most often hearing loss is diagnosed at the age of 6-10 years. Hearing loss in children begins at high frequencies, reaching a significant degree in air and bone conduction, passing from sound-conducting to sound-receiving hearing loss. Hearing loss can be one of the first symptoms of the disease and can precede urinary syndrome.
In 20% of cases, patients with Alport syndrome have changes in the eyes. The most common anomalies from the lens: spherofokiya, lentikonus anterior, posterior or mixed, a variety of cataracts. In families with Alport syndrome there is a significant incidence of myopia. A number of researchers constantly in these families note bilateral perimacular changes in the form of bright whitish or yellowish granulations in the area of the yellow body. They regard this symptom as a constant symptom, which has high diagnostic value in Alport syndrome. C. S. Chugh et al. (1993), in an ophthalmologic study, patients with alport syndrome showed a decrease in visual acuity in 66.7% of cases, a front lentikonus in 37.8%, spots on the retina in 22.2%, cataracts in 20%, keratoconus in 6 , 7%.
In some children with hereditary nephritis, especially in the formation of renal insufficiency, a significant lag in physical development is noted. As the progression of renal insufficiency develops hypertension. In children, it is more often detected in adolescence and in older age groups.
Characteristic is the presence in patients with hereditary nephritis of various (more than 5-7) stigmas of connective tissue dysembryogenesis. Among the connective tissue stigmas in patients the most common are hypertelorism of the eyes, high palate, bite abnormalities, abnormal shape of the auricles, curling of the little finger on the hands, "sandal-like cleft" on the feet. For hereditary nephritis is characterized by the same type of stigma dizembryogenesis within the family, as well as the high frequency of their spread among relatives of probands, through which the disease is transmitted.
In the early stages of the disease, an isolated decrease in the partial functions of the kidneys is revealed: the transport of amino acids, electrolytes, the concentration function, acidogenesis, later the changes concern the functional state of both the proximal and the distal nephron and are of the nature of combined partial disturbances. Reduction of glomerular filtration occurs later, more often in the adolescent period. As the hereditary nephritis progresses, anemia develops.
Thus, for hereditary nephritis, the staging of the disease is typical: first the latent stage or hidden clinical symptoms, manifested by minimal changes in the urinary syndrome, followed by the gradual decompensation of the process with the reduction of renal functions with manifest clinical symptoms (intoxication, asthenia, developmental deficiency, anemia). Clinical symptoms appear usually regardless of the stratification of the inflammatory reaction.
Hereditary nephritis can manifest in different age periods, which depends on the action of the gene, which until a certain time is in a repressed state.
Classification
There are three variants of hereditary nephritis
- I variant - is clinically manifested by nephritis with hematuria, hearing loss and eye damage. The course of nephritis is progressive with the development of CRF. The type of inheritance is dominant, linked to the X chromosome. Morphologically, there is a disturbance of the structure of the basal membrane, its thinning and cleavage.
- II variant-is clinically manifested by nephritis with hematuria without hearing loss. The course of nephritis is progressive with the development of chronic renal failure. The type of inheritance is dominant, linked to the X chromosome. Morphologically, thinning of the basal membrane of glomerular capillaries (especially laminadensa) is revealed.
- III option - benign family hematuria. The course is favorable, chronic renal failure does not develop. The type of inheritance is autosomal dominant or autosomal recessive. In the autosomal recessive type of inheritance, women have a more severe course of the disease.
Diagnosis of Alport syndrome
The following criteria are proposed:
- the presence in each family of at least two patients with nephropathy;
- hematuria as the leading symptom of nephropathy in the proband;
- at least one member of the family has a hearing loss;
- development of chronic kidney failure in one relative and more.
When diagnosing a variety of hereditary and congenital diseases, a comprehensive approach to the survey is of great importance, and first of all attention to the data obtained in the compilation of the pedigree of the child. The diagnosis of Alport syndrome is considered competent in cases when the patient has 3 out of 4 typical signs: the presence of hematuria in the family and chronic renal failure, the presence of neurosensory hearing loss in the patient, the pathology of vision, the detection of signs of splitting of the glomerular basement membrane with a change in its thickness in the electron microscopic characteristic of the biopsy specimen and uneven contours.
The examination of the patient should include clinical genetic methods of investigation; directed study of anamnesis of the disease; general examination of the patient taking into account diagnostic criteria. In the compensation stage, one can catch pathology only by focusing on such syndromes as the presence of hereditary complication, hypotension, multiple stigma of dysembryogenesis, changes in the urinary syndrome. In the stage of decompensation, there may be the appearance of estrarenal symptoms, such as severe intoxication, asthenia, lag in physical development, anemia, manifested and amplified with a gradual decrease in renal function. In most patients with a decrease in renal function, a decrease in the function of acido- and aminogenesis is observed; in 50% of patients notice a significant decrease in the secretory function of the kidneys; limiting the range of fluctuations in the optical density of urine; a violation of the rhythm of filtration, and then a decrease in glomerular filtration. The stage of chronic renal failure is diagnosed in patients with a 3-6 month and more elevated urea level in the blood serum (more than 0.35 g / l), a decrease in glomerular filtration to 25% of the norm.
Differential diagnosis of hereditary nephritis must be carried out primarily with the hematuric form of acquired glomerulonephritis. The acquired glomerulonephritis often has an acute onset, a period of 2-3 weeks after the infection, extrarenal signs, including hypertension from the first days (with hereditary nephritis, on the contrary, hypotension), a decrease in glomerular filtration at the onset of the disease, no violation of the partial tubular functions, then as with hereditary they are present. Acquired glomerulonephritis occurs with more pronounced hematuria and proteinuria, with increased ESR. The typical changes in the glomerular basement membrane characteristic of hereditary nephritis are of diagnostic significance.
Differential diagnosis of dysmetabolic nephropathy is carried out with chronic renal failure, the family clinically identifies multiple types of kidney disease, and maybe a spectrum of nephropathy from pyelonephritis to urolithiasis. Children often have complaints of abdominal pain and periodically with urination, in the urine sediment - oxalate.
If you suspect a hereditary nephritis patient should be sent to clarify the diagnosis in a specialized nephrology department.
What do need to examine?
What tests are needed?
Who to contact?
Treatment of Alport syndrome
In the regime provides for a restriction of large physical exertion, stay in the fresh air. The diet is high-grade, with sufficient content of high-grade proteins, fats and carbohydrates taking into account the function of the kidneys. Of great importance is the identification and rehabilitation of chronic foci of infection. From drugs, ATP, cocarboxylase, pyridoxine (up to 50 mg / day), carnitine chloride are used. The courses are held 2-3 times a year. When hematuria is prescribed phytotherapy - nettle, nettle, blackberry ash, yarrow.
In foreign and domestic literature there are reports of treatment with prednisolone and the use of cytostatics. However, the effect is difficult to judge.
In chronic renal failure, hemodialysis and kidney transplantation are used.
There are no methods of specific (effective pathogenetic) therapy of hereditary nephritis. All medical measures are aimed at preventing and slowing down the reduction of renal functions.
The diet should be balanced and high-calorie, taking into account the functional state of the kidneys. In the absence of violations of the functional state in the nutrition of the child should be a sufficient content of proteins, fats and carbohydrates. In the presence of signs of renal dysfunction, the amount of protein, carbohydrates of calcium and phosphorus should be limited, which delays the development of chronic kidney failure.
Physical stress should be limited, children are advised to refrain from doing sports.
Avoid contact with infectious patients, reduce the risk of developing acute respiratory infections. It is necessary to sanitize foci of chronic infection. Preventive vaccinations for children with hereditary nephritis are not carried out, vaccination is possible only according to epidemiological indications.
Hormonal and immunosuppressive therapy in hereditary nephritis is ineffective. There are indications of a certain positive effect (a decrease in the level of proteinuria and a slowdown in the progression of the disease) with long-term use of cyclosporine A and ACE inhibitors for many years.
In the treatment of patients using drugs that improve metabolism:
- pyridoxine - 2-3 mg / kg / day in 3 divided doses for 4 weeks;
- kokarboksilaza - 50 mg intramuscularly every other day, only 10-15 injections;
- ATP - 1 ml intramuscularly every other day, 10-15 injections;
- Vitamin A - 1000 U / year / day in 1 reception for 2 weeks;
- vitamin E - 1 mg / kg / day in 1 reception for 2 weeks.
Such therapy improves the general condition of patients, reduces tubular dysfunction, and is administered 3 times a year.
As an immunomodulator may be used levamisole - 2 mg / kg / day 2-3 times a week with intermissions between doses of 3-4 days.
To the researchers, hyperbaric oxygenation has a positive effect on the severity of hematuria and renal dysfunction.
The most effective method of treating hereditary nephritis is timely kidney transplantation. There is no recurrence of the disease in the transplant, in a small percentage of cases (about 5%), the development of nephritis in the transplanted kidney associated with antigens to the glomerular basement membrane is possible.
A promising area is prenatal diagnosis and genetic engineering therapy. Experiments on animals show a high efficiency of transferring normal genes responsible for the synthesis of a-chains of type IV collagen to the kidney tissue, after which synthesis of normal collagen structures is noted.
Forecast
The prognosis of hereditary nephritis is always serious.
Prognostically unfavorable criteria for the flow of hereditary nephritis are:
- male;
- early development of chronic renal failure in family members;
- proteinuria (more than 1 g / day);
- thickening of glomerular basal membranes according to microscopy;
- neuritis of the auditory nerve;
- deletion in the gene Co4A5.
The prognosis of benign family hematuria is more favorable.
Использованная литература