New publications
New findings contribute to a better understanding of the causes of Rett syndrome
Last reviewed: 02.07.2025

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.

Rett syndrome is a rare neurodevelopmental disorder for which there is currently no cure or good treatment. It causes severe physical and cognitive symptoms, many of which overlap with autism spectrum disorders.
Rett syndrome is caused by mutations in the MECP2 gene, which is highly expressed in the brain and appears to play an important role in maintaining the health of neurons. The gene is located on the X chromosome, and the syndrome primarily affects girls. To develop treatments for Rett syndrome, researchers want to better understand MECP2 and its functions in the brain.
Researchers, including Whitehead Institute co-founder Rudolf Jaenisch, have been studying MECP2 for decades, yet many basic facts about the gene remained unknown. The protein encoded by the gene, MECP2, is involved in gene regulation; it binds to DNA and influences the expression levels of various other genes, or the amount of protein they produce.
However, researchers did not have a complete list of genes affected by MECP2, and there was no consensus on how MECP2 affects these genes.
Early studies of MECP2 suggested that it was a repressor, reducing the expression of its target genes, but research by Jaenisch and others had previously shown that MECP2 also acts as an activator, increasing the expression of its targets — and that it may be an activator in the first place. Also unknown was MECP2’s mechanism of action, or what exactly the protein does to cause changes in gene expression.
Limitations in technology have prevented researchers from gaining clarity on these questions. But Yanish, his lab’s postdoc Yi Liu, and Yanish’s former lab member Anthony Flamier, now an assistant professor at the CHU Sainte-Justine research center at the Université de Montréal, have used cutting-edge techniques to answer these remaining questions about MECP2 and gain new insights into its role in brain health and disease.
Their results were published in the journal Neuron and the researchers also created an online repository of their MECP2 data, the MECP2-NeuroAtlas portal, as a resource for other researchers.
"I think this paper will fundamentally change people's understanding of how MECP2 causes Rett syndrome. We have a completely new understanding of the mechanism, and it may provide new avenues for developing treatments for the disease," says Janisch, who is also a professor of biology at MIT.
Deeper Understanding of MECP2 in the Brain
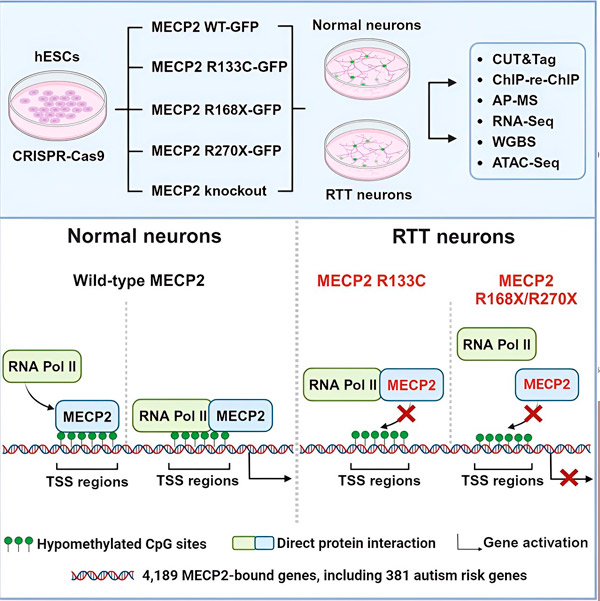
The researchers first created a detailed map of where MECP2 binds in human neuronal gene sequences, either within genes or in regulatory regions of DNA near them. They used an approach called CUT&Tag, which can pinpoint protein interactions with DNA with high precision.
The researchers found more than 4,000 genes associated with MECP2. They repeated their mapping in neurons with common MECP2 mutations associated with Rett syndrome to determine where MECP2 is depleted in the disease state.
Knowing which genes MECP2 binds to allowed Liu and Flamier to begin making connections between MECP2’s targets and brain health. They found that many of its targets are involved in the development and function of neuronal axons and synapses.
They also compared their list of MECP2 targets with the Simons Foundation Autism Research Initiative (SFARI) database of autism-associated genes and found that 381 genes in that database are MECP2 targets.
Source: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
These findings may help clarify the mechanisms underlying autism symptoms in Rett syndrome and provide a good starting point for investigating the possible role of MECP2 in autism.
"We have created the first integrated map of the MECP2 epigenome in health and disease, and this map can guide future research," Liu says. "Knowing which genes are targets of MECP2, and which genes are directly disrupted in the disease, provides a solid foundation for understanding Rett syndrome and asking questions about gene regulation in neurons."
The researchers also looked at whether MECP2 increased or decreased the expression of its target genes. Consistent with the history of MECP2 being identified by some as an activator and by others as a repressor, Liu and Flamier found examples where MECP2 played both roles.
However, while MECP2 is more often thought of as a repressor, Liu and Flamier found that it is mostly an activator— confirming previous findings by Jaenisch and Liu. One new experiment showed that MECP2 activates at least 80% of its targets, and another found that it activates up to 88% of its targets.
The map of target genes created by the researchers provided additional insight into MECP2's role as an activator. They found that for genes that MECP2 activates, it typically binds to a region of DNA upstream of the gene called the transcription start site.
This is the site where cellular machinery initiates the process of transcribing a gene into RNA, after which the RNA is translated into a functional protein, which is the product of gene expression. The presence of MECP2 at the transcription start site, where gene expression begins, is consistent with its role as a gene activator.
The researchers then set out to determine what role MECP2 plays in gene activation. They looked at what molecules MECP2 binds to at this site, in addition to DNA, and found that MECP2 interacts directly with a protein complex called RNA polymerase II (RNA Pol II). RNA Pol II is a key cellular machine that transcribes DNA into RNA. RNA Pol II can’t find genes on its own, so it requires a variety of cofactors, or protein collaborators, to help it do its job.
The researchers propose that MECP2 serves as one such cofactor, helping RNA Pol II initiate transcription at genes where MECP2 binds. Structural analysis of MECP2 has identified parts of the molecule that bind to RNA Pol II, and other experiments have confirmed that loss of MECP2 reduces the presence of RNA Pol II at appropriate transcription start sites, as well as the expression levels of target genes.
This suggests that Rett syndrome may be caused by decreased transcription of genes targeted by MECP2 due to MECP2 mutations that prevent it from binding to RNA Pol II or binding to DNA. Consistent with this idea, most common MECP2 mutations associated with disease are truncations: mutations in which part of the protein is missing, which may alter the interaction between MECP2 and RNA Pol II.
The researchers hope that their findings will not only change our understanding of MECP2, but that a deeper and broader understanding of how MECP2 influences brain development and function could lead to new insights that will help people with Rett syndrome and related disorders, including autism.
"This project is a great example of the collaborative nature of the Janisch lab," says Flamier. "Rudolf and I had a specific problem related to Rett syndrome, and I had experience with the CUT&Tag technology, which could solve the problem. Through discussion, we realized that we could combine our efforts, and now we have a great repository of information about MECP2 and its links to disease."